
Translate this page into:
Idiopathic pulmonary fibrosis in Soweto, South Africa: A descriptive study
*Corresponding author: Wesley Mark Aitchison, Department of Internal Medicine, Chris Hani Baragwanath Academic Hospital, Soweto, South Africa. drwma@icloud.com
-
Received: ,
Accepted: ,
How to cite this article: Aitchison WM, Van Blydenstein SA, Wong M. Idiopathic pulmonary fibrosis in Soweto, South Africa: A descriptive study. J Pan Afr Thorac Soc 2023;4:81-9.
Abstract
Objectives:
Idiopathic pulmonary fibrosis (IPF) is a specific form of age-related fibroproliferative interstitial pneumonia that is chronic, progressive, and carries a poor prognosis, with median survival of just 2.5–3.5 years from diagnosis. The exact etiology is unknown, but smoking is known to be risk factor. Symptoms and signs include progressive dyspnoea, cough, inspiratory “Velcro” crackles, and clubbing. At present, treatment options are limited; but include pulmonary rehabilitation, long-term domiciliary oxygen therapy, and the conditionally recommended pharmacological therapies pirfenidone and nintedanib. This study sought to describe the cohort of patients that attended the respiratory outpatient services at a tertiary-level hospital in South Africa during the period 2007– 2016. To the best of the authors’ knowledge, this is the first such descriptive study performed in Africa.
Materials and Methods:
This was a retrospective, descriptive, and record review, that included patients ≥18 years of age who fulfilled 2011 ATS/ERS/JRS/ALAT diagnostic criteria for IPF.
Results:
Data from 74 patients were used for analysis in this study, of which 60.8% were female. The mean age standard deviation was 64.4 (10.9) years and the majority (79.7%) were Black. Over half of the patients (40/74, 54.1%) were current or previous smokers, although there was no correlation between smoking history and age or baseline pulmonary function testing. All patients reported dyspnea, which was modified Medical Reseach Council (mMRC) Grade 3 or 4 in 80% of patients. High resolution computed tomography chest was reported as radiological usual interstitial pneumonia (UIP) in 72 patients (97.3%) and three patients underwent lung biopsy, all of which showed a UIP pattern. Fifty-eight patients (78.4%) had spirometry results available, with median forced vital capacity 67.3% of predicted; this was significantly higher in females. Median transfer factor of the lung for carbon monoxide was 39% predicted. Twenty-five patients (33.8%) received corticosteroids, of whom five (6.8%) received the prednisone-azathioprine-N-acetylcysteine regime. Three patients (4.1%) received nintedanib; two of whom showed slowing of decline in lung function, although no significant symptomatic improvement was reported. Mean duration of follow-up was 13.3 months, although females had significantly longer duration of follow-up than males.
Conclusion:
Despite a fairly small sample size and retrospective nature, this study contributes to the body of literature on IPF and highlights the need for additional studies in developing countries, particularly in Africa.
Keywords
Idiopathic pulmonary fibrosis
IPF
Interstitial lung disease
ILD
Diffuse parenchymal lung disease (DPLD)

INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a specific form of age-related fibroproliferative interstitial pneumonia that is chronic, progressive, and carries a poor prognosis.[1] Although rare, IPF is the most common of the idiopathic interstitial pneumonias.[2,3] The annual incidence of IPF is estimated to be 4.6–16.3 cases/100 000 people and appears to be increasing,[4] although incidence in individuals 75 years and older may be closer to 80/100 000 people.[5] The exact etiology of IPF is unknown, but several potential risk factors have been identified; smoking, exposure to metal and wood dust, gastro-esophageal reflux disease, certain viral infections, exposure to livestock, certain occupations, and various genetic predispositions. The pathogenesis of IPF is complex and poorly understood and is thought to be initiated by an unknown inciting factor or exposure in a genetically susceptible individual, with dysfunction of fibroblasts and epithelial cells, succeeded by progressive fibrosis.[6-10] Smoking in particular is a strong risk factor for the development of IPF.[11,12]
Typical symptoms on presentation include gradual onset of dyspnea on exertion and non-productive cough, usually progressing over several months. Extrapulmonary manifestations may be found only in genetic forms of IPF. Thorough history-taking is critically important to establish other possible etiologies of interstitial lung disease (ILD). Clinical features include bibasal, fine inspiratory crackles (often termed “Velcro” crackles), and clubbing (in 25–75% of patients). If presenting late, patients may have features of pulmonary hypertension, corpulmonale, and right heart failure.[1,4,13,14]
The diagnosis of IPF requires the exclusion of other known causes of ILD and a typical usual interstitial pneumonia (UIP) pattern on high resolution computed tomography (HRCT) in patients not subjected to lung biopsy.[15] Pulmonary function tests in patients with IPF usually show a restrictive pattern, with a reduced forced vital capacity (FVC) and normal to high forced expiratory volume in 1 s (FEV1) to FVC ratio, low residual volume (RV) and total lung capacity (TLC), and a reduced transfer factor of the lung for carbon monoxide (TLCO), which typically declines with disease progression.[1,16,17]
Treatment options for IPF are limited, with previously-used treatment regimens being shown to be ineffective or even harmful, such as azathioprine-prednisone-Nacetylcysteine.[18,19] Supportive care including pulmonary rehabilitation, education, vaccination, and long-term domiciliary oxygen therapy (LTDOT) are currently recommended, and form a critical component of the management of IPF, and smoking cessation should be encouraged.[1,20] At present, the only conditionally recommended pharmacological therapies are pirfenidone and nintedanib, while anti-acid agents may be considered in appropriate patients.[1,4,18] These drugs are both very costly, but do seem to improve overall quality of life.[1,21] There does not appear to be a significant difference between pirfenidone and nintedanib in terms of reduction in rate of FVC decline reduction or mortality when compared.[22]
The clinical course of IPF is somewhat heterogeneous, with median survival just 2.5–3.5 years from diagnosis, although up to one quarter of patients may live beyond 10 years.[4] Up to 20% of patients experience acute exacerbations, which carry a mortality rate of at least 50%, even in developed countries.[23] The median survival following an acute exacerbation is 3–4 months.[24]
At present, there are very limited South African data available regarding the incidence, presentation, and risk factors in this population, and none since the introduction of the 2011 ATS/ERS/JRS/ALAT diagnostic criteria[15] (see below). As such, this study aimed to describe the cohort of patients that attend or have attended the respiratory outpatient services at a South African tertiary-level hospital.
The 2011 ATS/ERS/JRS/ALAT diagnostic criteria are as follows: [15]
Exclusion of other known causes of ILD (e.g., domestic and occupational environmental exposures, connective tissue disease, and drug toxicity) AND
The presence of a UIP pattern on HRCT in patients not subjected to surgical lung biopsy OR
Specific combinations of HRCT and surgical lung biopsy patterns in patients subjected to surgical lung biopsy, require multidisciplinary discussion.
MATERIALS AND METHODS
Study design and setting
A retrospective, descriptive, and record review was undertaken at the Respiratory Outpatients Department at Chris Hani Baragwanath Academic Hospital. This tertiary-level academic institution in Soweto, South Africa, is affiliated with the University of the Witwatersrand.
Data collection and sample selection
Data from patient records were collected, including demographics, clinical, laboratory, and radiologic findings, lung function tests, treatment, date of last follow-up visit and, where applicable and available, date of death. A pre-existing database of patients was used to identify patients suspected or diagnosed with IPF during the study period (2007–2016); 115 such patients were identified. The following criteria were then applied:
Inclusion criteria: Patients aged ≥ 18 years who had been evaluated by the Division of Pulmonology at Chris Hani Baragwanath Academic Hospital and met the 2011 ATS/ ERS/JRS/ALAT diagnostic criteria for IPF.[20]
Exclusion criteria: files in which it is documented that the patient requested their information not be used for studies.
Descriptive statistics were used to describe baseline demographics and clinical profile of study participants. Categorical variables were described using frequencies and percentages, while skewed continuous variables were described using the median and interquartile range. Bivariate analysis for categorical variables was assessed using the Pearson’s Chi-squared test, otherwise Fisher’s exact for sparse data. The Wilcoxon rank-sum test was used to evaluate equality of medians between groups where applicable. Linear regression models determined factors associated with decline in predicted FVC and FEV1 values (%) over time. Body mass index (BMI) was calculated using weight and height where documented and defined as noted in [Table 1].
| Variable | n(%) |
|---|---|
| Sex | |
| Female | 45 (60.8) |
| Male | 29 (39.2) |
| Age | |
| Mean, SD (Range) | 64.4, 10.9 (33–86) |
| <60 | 18 (24.3) |
| 60–70 | 31 (41.9) |
| ≥70 | 25 (33.8) |
| Ethnicity | |
| Black | 59 (79.7) |
| White | 8 (10.8) |
| Indian | 6 (8.1) |
| Colored | 1 (1.4) |
| HIV status | |
| Positive | 1 (1.4) |
| Negative | 45 (60.8) |
| Unknown | 28 (37.8) |
| Smoking status | |
| Never smoked | 32 (43.2) |
| Current smoker | 5 (6.8) |
| Pack years: Mean (SD) | 50.4 (27.1) |
| Previous smoker | 35 (47.3) |
| Pack years: Mean (SD) | 28.0 (16.7) |
| Unknown | 2 (2.7) |
| BMI (kg/m2) | |
| <18.5 (underweight) | 4 (5.4) |
| 18.5–24.9 (normal weight) | 20 (27.0) |
| ≥25.0 (overweight) | 26 (35.1) |
| Undocumented | 24 (32.4) |
| TB history | |
| Never had TB | 57 (77.0) |
| Current pulmonary TB | 7 (9.5) |
| Previous pulmonary TB | 10 (13.5) |
| Documented exposures | |
| Construction/Dust | 2 (2.7) |
| Mining/Asbestos | 4 (5.4) |
| Paint and/or cleaning chemicals | 3 (4.1) |
| Comorbidities | |
| Hypertension | 35 (47.3) |
| COPD | 4 (5.4) |
| Other comorbidities | |
| Asthma | 1 (1.4%) |
| Cardiomyopathy/ischemic heart disease | 7 (9.5%) |
| Diabetes mellitus | 9 (12.1) |
| Gastro-esophageal reflux disease | 2 (2.7) |
| Lung Cancer | 1 (1.4) |
| Pulmonary thrombo-embolic disease | 2 (2.7) |
| Others (aortic stenosis, aortic aneurysm, atrial fibrillation, chronic kidney disease, epilepsy, fatty liver disease, gout, hypothyroidism, osteoarthritis, and prostate cancer) | 13 (17.6) |
| None known | 25 (33.8) |
SD: Standard deviation, HIV: Human immunodeficiency virus, COPD: Chronic obstructive pulmonary disease, TB: Tuberculosis, BMI: Body mass index, n: number
Measure outcomes
To describe the demographics, presentation symptoms, and clinical features of patients in the sample population
To determine any identifiable risk factors and associations present in this population, including laboratory studies performed
To determine the presentation and subsequent lung functions, including but not limited to forced vital capacity, TLC, and TLCO in this population
To determine treatment regimens used in this population, and the effect (if any) on symptoms and lung function
Where documented, to determine mean time from symptom development to death, as well as from diagnosis to death, in patients who have died, and to thereby demonstrate if diagnosis was delayed.
RESULTS
Patient characteristics
Of the 115 patients identified for inclusion, 3 (2.6%) were excluded due to lack of records, and a further 38 (33.0%) were excluded as they did not meet the 2011 ATS/ERS/JRS/ALAT diagnostic criteria,[20] thus resulting in 74 patients’ data being used for analysis. The demographic and clinical characteristics of these patients at presentation are summarized in [Table 1]. The mean (range, SD) age was 64.4 years (33–86, 10.9), and 45 patients were female (60.8%). Difference in age between males and females was not statistically significant (P = 0.17). Fifty-nine (79.7%) of the patients were of Black ethnicity. Mean BMI among females was 26.8 kg/m2, versus 24.6 kg/m2 among males (P = 0.07). Mean time from symptom onset to diagnosis was 17.4 months (range 1.6–86.4 months) and was not significantly different between females and males (17.0 vs. 18.0 months, P = 0.41), nor between Black and non-Black ethnic groups (18.6 vs. 12.9 months, P = 0.09).
One patient (1.4%) was Human Immunodeficiency Virus (HIV)-positive on anti-retroviral therapy, although HIV status was not determined in 38.7%. Forty patients (54.0%) were either current or previous smokers, four of whom had been clinically diagnosed with chronic obstructive pulmonary disease (COPD). Males were significantly more likely to be current or previous smokers than females (75.8% vs. 40.0%, P = 0.007). White patients were significantly more likely to be current or previous smokers than Black patients (87.5% vs. 49.2%, P = 0.007). Hypertension was the most common comorbidity, present in 35 patients (47.3%), followed by diabetes mellitus in 9 patients (12.1%).
Features at presentation and risk factors
A summary of symptoms and clinical signs at presentation is presented in [Figure 1]. All patients reported dyspnea on presentation, and most patients (79.7%) presented with either mMRC dyspnea score 3 or 4. Cough was present in all but one patient. Seventy-two patients (97.3%) had inspiratory “Velcro” crackles on presentation, while 35 patients (42.3%) had clubbing. The chest radiograph findings of 70 patients were documented, of which 70 (100%) showed bilateral reticular infiltrates. Two patients (2.7%) also had radiographic hilar lymphadenopathy, while an additional 2 patients (2.7%) had features of bronchiectasis documented. Neither of these patients was known to have had previous tuberculosis (TB). One patient (1.4%) had a parenchymal mass visible on chest radiography and HRCT chest, suspected of being primary lung cancer. HRCT chest was reported as radiological UIP in 72 patients (97.3%), (including the abovementioned patients with bronchiectasis and parenchymal mass), while 2 patients (2.7%) were reported as radiological nonspecific interstitial pneumonia (NSIP). Additional findings included COPD in two patients (2.7%). One patient had a history of asbestos exposure, but no asbestos-related pleural abnormalities, suggesting that the diagnosis was more likely IPF than asbestosis. Three patients (4.1%) underwent lung biopsies, two of which were performed on the patients reported to have NSIP on HRCT. All three lung biopsies showed UIP. Baseline pulmonary function testing (PFT) (where available/ documented) is shown in [Table 2]. Fifty-eight patients (78.4%) had spirometry results documented. Percentage of predicted FVC was significantly higher amongst females (median 72.0 vs. 62.0, P = 0.04), as was percentage of predicted FEV1 (median 83.0 vs. 61.0, P = 0.026). FEV1/FVC ratio was not significantly different between males and females (median 83.3 vs. 84.9, P = 0.89). Results of full PFT were available in 32 patients (43.2%), which revealed no significant differences in percentage of predicted values in TLC, RV, or TLCO between males and females. There was also no significant correlation between pack year history and baseline pulmonary function tests [Figure 2]. Using available data, the gender-age-physiology (GAP) score[25] could be calculated for 32 patients. The scores and corresponding stages are presented in [Figure 3]. The majority of females were GAP Stage II on presentation (14 patients, 63.6%), while 5 male patients (50%) were Stage II (P = 0.24).

- Symptoms and signs at presentation (n=74).
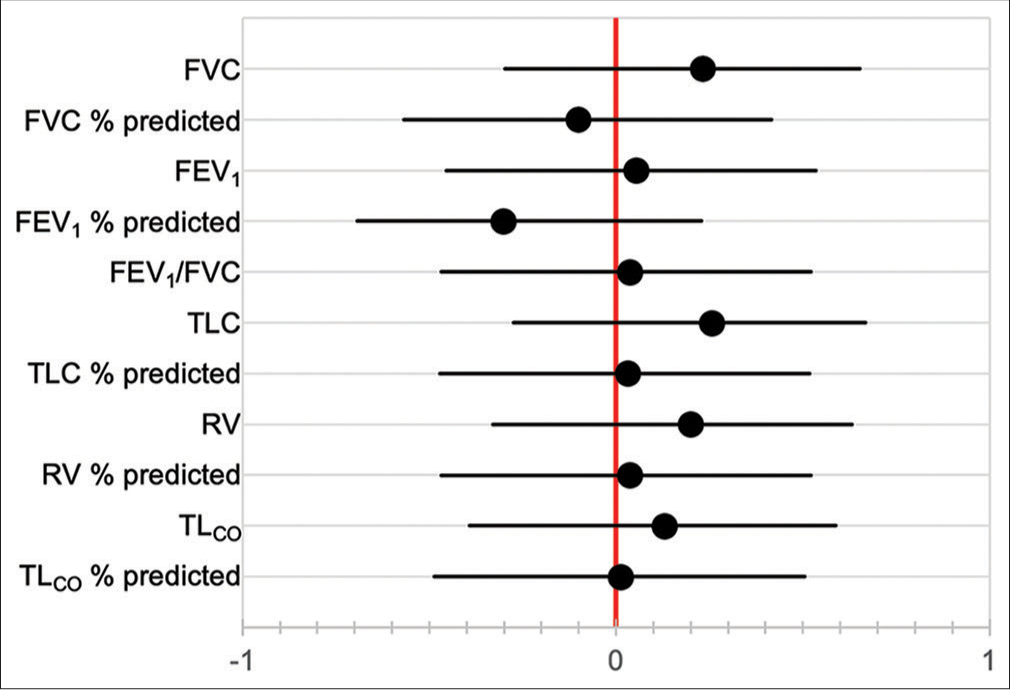
- Correlation coefficients between smoking pack year history and baseline pulmonary function tests. FVC: forced vital capacity, FEV1: forced expiratory volume in 1 second, TLC: total lung capacity, RV: residual volume, TLCO: transfer factor of the lung for carbon monoxide.
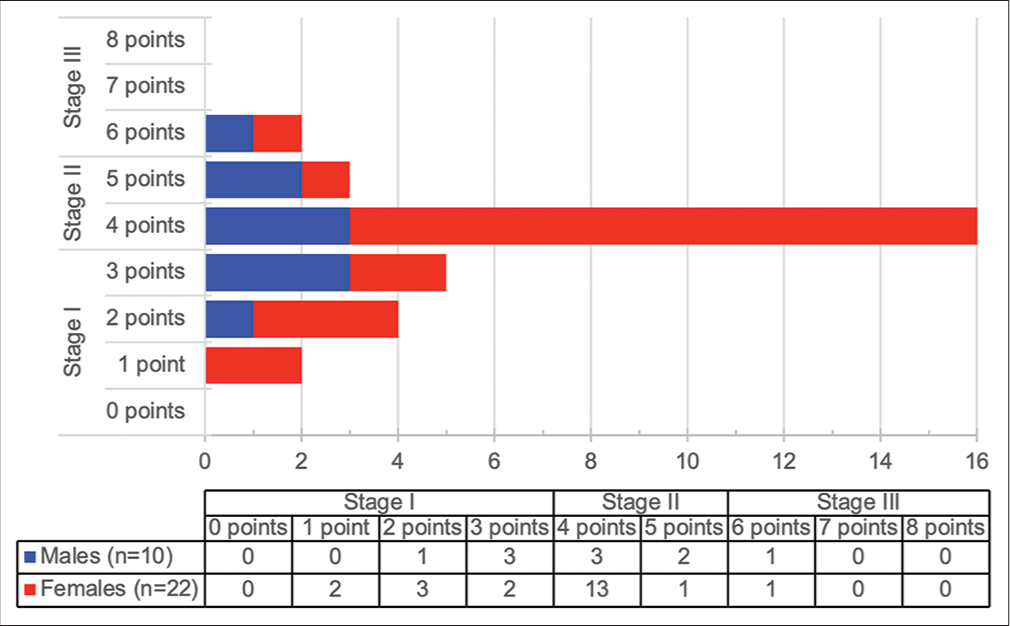
- Gender-age-physiology score and stage (n=32).
| Variable | All | Females | Males | P-value |
|---|---|---|---|---|
| FVC (L) (median, range) | 1.8 (1.3–2.1) | 1.6 (1.2–2.0) | 2.1 (1.8–2.4) | |
| [n=58] | [n=35] | [n=23] | ||
| FVC (% predicted) | ||||
| Median (IQR) | 67.3 (54.0–89.0) | 72.0 (55.0–96.0) | 62.0 (52.9–73) | 0.040* |
| <80% (n[%]) | 39 (67.2%) | 20 (57.1%) | 19 (82.6%) | |
| ≥80% (n[%]) | 19 (32.8%) | 15 (42.9%) | 4 (17.4%) | |
| FEV1(L) (median, range) | 1.5 (1.1–1.8) | 1.3 (1.1–1.6) | 1.7 (1.3–2.0) | |
| [n=57] | [n=34] | [n=23] | ||
| FEV1(% predicted) | ||||
| Median (IQR) | 71.0 (52.0–91.0) | 83.0 (54.0–98.0) | 61.0 (51.3–82.0) | 0.026* |
| <80% (n[%]) | 32 (56.1%) | 15 (44.1%) | 17 (73.9%) | |
| ≥80% (n[%]) | 25 (43.9%) | 19 (55.9%) | 6 (26.1%) | |
| FEV1/FVC ratio (%) | 83.6 (76.8–88.0) | 83.3 (76.8–87.0) | 84.9 (73.0–88.6) | 0.890 |
| [n=57] | [n=34] | [n=23] | ||
| TLC (L) (median, range) | 2.9 (2.1–3.2) | 2.6 (2.0–3.1) | 3.2 (3.0–3.9) | |
| [n=32] | [n=22] | [n=10] | ||
| TLC (% predicted) | ||||
| Median (IQR) | 59.0 (51.0–77.0) | 60.0 (51.0–78.0) | 59.0 (54.0–67.0) | 0.624 |
| <80% (n[%]) | 25 (78.1%) | 17 (77.3%) | 8 (80.0%) | |
| ≥80% (n[%]) | 7 (21.9%) | 5 (22.7%) | 2 (20.0%) | |
| RV (L) (median, range) | 1.1 (0.5–1.4) | 0.9 (0.5–1.4) | 1.3 (0.9–1.7) | |
| [n=32] | [n=22] | [n=10] | ||
| RV (% predicted) | ||||
| Median (IQR) | 58.0 (36.5–81.5) | 58.0 (30.0–90.0) | 59.0 (49.0–78.0) | 0.703 |
| <80% (n[%]) | 24 (68.0%) | 16 (72.7%) | 8 (80.0%) | |
| ≥80% (n[%]) | 8 (32.0%) | 6 (27.3%) | 2 (20.0%) | |
| TLCO(mmol/min/mm Hg) (median, range) | 7.5 (4.2–10.0) | 5.5 (4.1–8.0) | 9.5 (7.5–10.2) | |
| [n=31] | [n=21] | [n=10] | ||
| TLCO(% predicted) | ||||
| Median (IQR) | 39.0 (25.0–47.0) | 29.0 (25.0–47.0) | 40.5 (29.0–45.0) | 0.323 |
| <80% (n[%]) | 30 (93.8%) | 21 (100.0%) | 9 (90.0%) | |
| ≥80% (n[%]) | 1 (3.2%) | 0 (0.0%) | 1 (10.0%) |
IPF: Idiopathic pulmonary fibrosis, IQR: Inter quartile range, FVC: Forced vital capacity, FEV1: Forced expiratory volume in 1 s, TLC: Total lung capacity, RV: Residual volume, TLCO: Transfer factor of the lung for carbon monoxide. *Denotes statistical significance (P<0.05)
Echocardiographic findings were documented in 19 patients (25.7%), although only 14 of these included pulmonary artery pressures (PAP), and 18 recorded left ventricular ejection fraction (LVEF). PAP was elevated in all 14 patients (range 34–110, median 53.5 mm Hg), and clinical features of pulmonary hypertension were recorded in 13 of these patients (92.9%). There was no significant difference between PAP in males and females (P = 0.37). The median ejection fraction was 65.5% (interquartile range 61–67.8%). Two patients had a LVEF < 50%; these patients both had documented cardiomyopathies.
Room air arterial blood gases were recorded in 32 patients (43.2%); the results of these can be found in [Table 3]. On formal laboratory testing, 56 patients (75.7%) had full blood count results documented, which revealed a mean hemoglobin concentration of 14.0 g/dL in females and 15.5 g/dL in males (P = 0.003). Other blood count parameters were not significantly different between males and females. Urea, electrolytes, and creatinine were available in 59 patients (79.7%); these results were also not significantly different between males and females. Serological studies including anti-nuclear antibody (ANA), rheumatoid factor, and serum angiotensin converting enzyme were used to screen for alternate causes of ILD.
| pH | pO2 (mmHg) | pCO2 (mmHg) | Oxygen saturation (%) | Base excess (mmol/ℓ) | HCO3−(mmol/ℓ) | |
|---|---|---|---|---|---|---|
| Median | 7.42 | 50.2 | 35.95 | 87 | −0.3 | 23.5 |
| Inter quartile range | 7.40–7.44 | 47.4–57.5 | 32.0–40.0 | 81.6–90.0 | −2.2–1.4 | 21.0–25.8 |
IPF: Idiopathic pulmonary fibrosis, pO2: partial pressure of oxygen, pCO2: partial pressure of carbon dioxide
ANA positivity was found in six patients (8.1%), all of whom were female, with no patients have a titer >1:160. Given the increasing prevalence of ANA positivity with age in the general population, this is not an unexpected finding.[26] These patients had no other documented symptoms or signs suggestive of autoimmune disease, and on this basis, rheumatological disease was excluded clinically.
Treatment regimens used
Corticosteroids were used in 25 patients (33.8%), with median prednisone dosage of 30 mg daily (range 5–60 mg) and median duration of 3 months (range 1–32 months). Two patients (8% of those treated with corticosteroids) developed vertebral compression fractures; these patients had received steroids for ≥ 30 months each. One further patient (4%) developed an upper gastrointestinal bleed after being treated for 16 months. One patient showed some symptomatic and spirometric improvement with corticosteroids, all others showed no improvement.
Ten patients (13.5%) received azathioprine, at a median dose of 75 mg daily (range 50–150 mg), and median duration of 3 months (range 1–33 months). These patients showed no improvement in symptoms with treatment. N-acetylcysteine (NAC) was used in five patients (6.8%); these patients all received the prednisone-azathioprine-NAC regimen, for between one and 6 months (median 3 months). All five patients were treated between 2009 and 2011. The total daily dosage range of N-acetylcysteine was 600–1800 mg. No change in condition was reported in patients receiving NAC. Proton pump inhibitors or H1-receptor blockers were used in 10 patients (13.5%). One patient received methotrexate at 7.5 mg weekly (in addition to corticosteroids).
Nintedanib was available through a compassionate use program for a limited period and was prescribed in three patients (4.1%), for a median duration of 29 months (range 8–35 months). Dosage used was 100 mg twice daily in one patient and 150 mg twice daily in the other two. Two patients receiving nintedanib showed slowing of decline in their lung function, although no significant symptomatic improvement was reported. No objective assessment could be made in the third patient as there were insufficient serial lung function tests performed for comment. This patient developed an acute exacerbation soon after nintedanib was initiated. One patient developed Clostridioides difficile-associated diarrhea. Adverse effects did not limit the use or treatment duration of nintedanib in any of the patients. Of the patients on nintedanib, one was still alive at the time of data collection. One patient died 8 months after starting nintedanib, and the other died after 4.5 years. No patients received pirfenidone. LTDOT was used in 36 patients (48.6%).
Follow-up and outcomes
Mean duration of follow-up was 13.3 months, with females having significantly longer follow-up durations than males (17.9 vs. 6.6 months, P < 0.001) [Figure 4]. Indian patients had significantly shorter mean duration of follow-up than Black patients (8.2 vs. 12.5 months, P = 0.006) and White patients (8.2 vs. 14 months, P = 0.02), while no significance was found between Black and White patients (P = 0.27). At the time of data collection, four patients were still alive (5.4%), 63 were lost to follow-up (85.1%), and seven were known to have died (9.5%). As noted in [Table 4], no significant differences between the different outcome groups were found in time from symptom development to first consultation or in time from symptom development to diagnosis.
| Outcome (at time of data collection) | n(%) | Median time from symptom development to first consultation (IQR) (months) | Median time from symptom development to diagnosis (IQR) (months) |
|---|---|---|---|
| Alive | 4 (5.4%) | 7.4 (3.2–12.5) | 9.3 (3.6–24.1) |
| Lost to follow-up | 63 (85.1%) | 9.8 (4.0–24.1) | 11.7 (4.8–24.1) |
| Dead | 7 (9.5%) | 7.1 (4.5–24.1) | 7.4 (6.3–24.1) |
| P-value | 0.774 | 0.951 |
IPF: Idiopathic pulmonary fibrosis, IQR: Inter quartile range
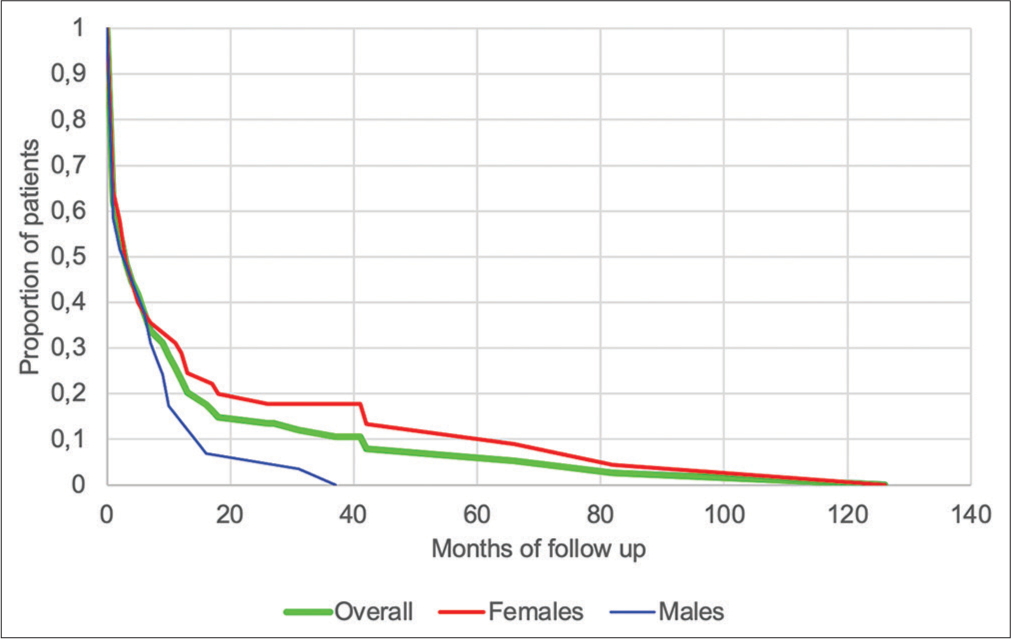
- Duration of follow-up of patients diagnosed with idiopathic pulmonary fibrosis (n=74).
Baseline PFT was not significantly correlated with duration of follow-up [Figure 5], and neither were baseline pO2 nor pCO2. Pack year history was also not associated with duration of follow-up (HR 1.004, 95% CI 0.988–1.020). Of the seven patients known to have died, three died at home, while four died in hospital. Cause of death was documented in only one patient, which was an acute exacerbation of IPF. Factors correlated with decline in FVC over time are shown in [Figure 6], although only 12 patients had serial PFT available for analysis. The factors most strongly correlated with decline in FVC were previous smoking history (P < 0.001), current smoking (P = 0.003), Black race (P = 0.039), and age (P = 0.044).
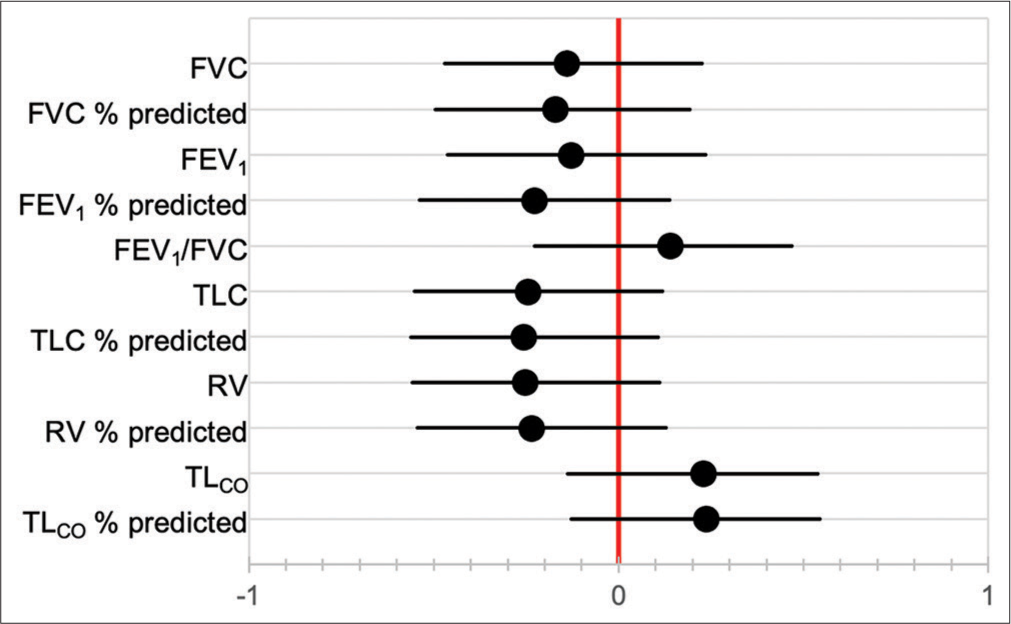
- Correlation coefficients between baseline pulmonary function tests and duration of follow-up.
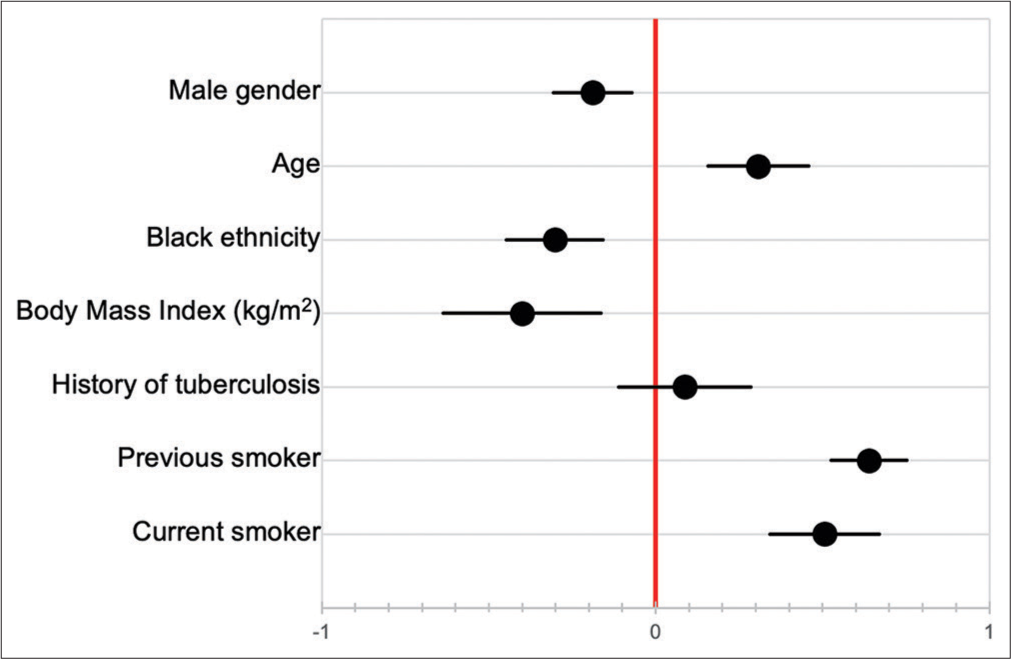
- Factors correlated with decline in forced vital capacity over time.
DISCUSSION
To the best of the authors’ knowledge, this is the first descriptive study of IPF in South Africa and Africa using the current definition, and included 74 patients. In keeping with existing literature,[15] the median age of our patient cohort was 64.4 years. The majority of our patients were female (60.8%), which conflicts with some existing data;[27,28] this may, however, be due to sampling bias, cultural differences, and different health-seeking behavior between sexes. Studies from India also found a female predominance, suggesting that epidemiology may differ in developing countries.[29,30] Our cohort was predominantly Black (79.7%); this was expected given the location and population served by Chris Hani Baragwanath Academic Hospital. The most common comorbidities were hypertension and diabetes mellitus, which was also expected given the age of patients in this cohort. Gastro-esophageal reflux disease was diagnosed in only 2.7% of our patients, although antacid therapy was used in 13.5%.
In this cohort, there was no difference in age of presentation between patients with a history of previous or current smoking, and those without; this is in contrast to some evidence that has shown that smokers may develop IPF at a younger age.[31,32] Smoking history (previous or current) did not correlate with baseline pulmonary function tests, although it was positively correlated with more rapid decline in FVC over time (correlation coefficient of 0.64 and 0.51 in previous and current smokers, respectively). Although limited by a relatively small sample size, this may either suggest that smoking is less of a risk factor in our patients, or more likely that other environmental factors such as exposure to passive smoking, biomass fuel, or environmental pollutants are important risk factors in the context of a developing country. No other risk factors could be defined in this study. Limited data prevented meaningful comparison of decline in FVC over time between races. Given conflicting existing data in this regard,[33,34] this is an area requiring further study, particularly in the South African context.
Patients universally presented with dyspnea, the majority of which were mMRC scores of 3 or 4. This, in combination with 71.6% of patients having clinical features of pulmonary hypertension, suggests advanced disease at presentation. Almost all patients had cough and inspiratory crackles, while clubbing was present in 42.3% of patients. The mean time from symptom development to diagnosis was 17.4 months. These findings are similar to those found in existing articles from both developing and developed countries.[13,14,35-37] While baseline FVC and FEV1 in this cohort were similar to those found by Collard et al,[37] RV and TLCO were markedly lower in our patients. This adds further evidence to the presence of advanced disease on presentation.
Pharmaceutical treatment regimens used in our patients included corticosteroids, prednisone-azathioprine-NAC, and nintedanib. LTDOT was used in almost half of patients. Too few data were available to analyze impact of these therapies on survival, as the majority of our patients were lost to follow up. There are several possible reasons for this, including death, disability, relocation, or socioeconomic difficulties in accessing further health-care services. Patients in our cohort also did not receive pulmonary rehabilitation or psychological support; implementation of such services would likely be of benefit to patients.
In addition to a fairly small sample size, a major limitation of this study was unavailability of some data, as it relied on a retrospective review of files. This resulted in limited data analyses, especially with regard to changes in PFTs over time and survival. Additional limitations include erratic and limited drug supply and lung function testing often being unavailable due to equipment breakdown, consumable shortages, and staffing problems.
CONCLUSION
Despite its limitations, this study contributes to the body of literature regarding IPF as the first descriptive study in Africa and highlights the need for additional work in understanding how IPF may present and progress differently in our setting. We found a female predominance, and no correlation between smoking history and age or baseline PFT, as well as greater decline in FVC over time in Black patients. These findings highlight the possibility of a different epidemiological pattern and clinical course in South African patients.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Recommendations for the management of idiopathic pulmonary fibrosis in South Africa: A position statement of the South African Thoracic Society. J Thorac Dis. 2016;8:3711-9.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157-79.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic pulmonary fibrosis: Evolving concepts. Mayo Clin Proc. 2014;89:1130-42.
- [CrossRef] [PubMed] [Google Scholar]
- Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810-6.
- [CrossRef] [PubMed] [Google Scholar]
- Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015;165:48-60.
- [CrossRef] [PubMed] [Google Scholar]
- Nonresolving fibrotic disorders: Idiopathic pulmonary fibrosis as a paradigm of impaired tissue regeneration. Am J Med Sci. 2011;341:431-4.
- [CrossRef] [PubMed] [Google Scholar]
- Extracellular matrix in normal and fibrotic human lungs. Am Rev Respir Dis. 1985;131:281-9.
- [Google Scholar]
- Analysis of cellular and protein content of broncho-alveolar lavage fluid from patients with idiopathic pulmonary fibrosis and chronic hypersensitivity pneumonitis. J Clin Invest. 1977;59:165-75.
- [CrossRef] [PubMed] [Google Scholar]
- Cigarette smoking aggravates bleomycin-induced experimental pulmonary fibrosis. Toxicol Lett. 2019;303:1-8.
- [CrossRef] [PubMed] [Google Scholar]
- Smoking-related idiopathic interstitial pneumonia: A review. Respirology. 2016;21:57-64.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and classification of idiopathic pulmonary fibrosis. Autoimmun Rev. 2014;13:508-12.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44-68.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary function testing in interstitial lung diseases. Respiration. 2004;71:209-13.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrotic idiopathic interstitial pneumonia: The prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168:531-7.
- [CrossRef] [PubMed] [Google Scholar]
- An official ATS/ERS/JRS/ALAT clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3-19.
- [CrossRef] [PubMed] [Google Scholar]
- Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968-77.
- [CrossRef] [PubMed] [Google Scholar]
- An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788-824.
- [CrossRef] [PubMed] [Google Scholar]
- Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382-92.
- [CrossRef] [PubMed] [Google Scholar]
- Systematic review and network meta-analysis of approved medicines for the treatment of idiopathic pulmonary fibrosis. J Drug Assess. 2019;8:55-61.
- [CrossRef] [PubMed] [Google Scholar]
- The burden of idiopathic pulmonary fibrosis: An unmet public health need. Respir Med. 2014;108:955-67.
- [CrossRef] [PubMed] [Google Scholar]
- Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur Respir J. 2011;37:356-63.
- [CrossRef] [PubMed] [Google Scholar]
- A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684-91.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and sociodemographic correlates of antinuclear antibodies in the United States. Arthritis Rheum. 2012;64:2319-27.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic pulmonary fibrosis in united states automated claims. Incidence, prevalence, and algorithm validation. Am J Respir Crit Care Med. 2015;192:1200-7.
- [CrossRef] [PubMed] [Google Scholar]
- Patient gender bias on the diagnosis of idiopathic pulmonary fibrosis. Thorax. 2020;75:407-12.
- [CrossRef] [PubMed] [Google Scholar]
- Retrospective study of interstitial lung disease in a tertiary care centre in India. Indian J Chest Dis Allied Sci. 2010;52:207-11.
- [CrossRef] [PubMed] [Google Scholar]
- Spectrum and diagnosis of idiopathic pulmonary fibrosis. Indian J Chest Dis Allied Sci. 2004;46:23-6.
- [Google Scholar]
- Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res. 2017;18:160.
- [CrossRef] [PubMed] [Google Scholar]
- Underlying and immediate causes of death in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. 2018;18:69.
- [CrossRef] [PubMed] [Google Scholar]
- Racial and ethnic disparities in idiopathic pulmonary fibrosis: A UNOS/OPTN database analysis. Am J Transplant. 2006;6:2436-42.
- [CrossRef] [PubMed] [Google Scholar]
- Ethnic and racial differences in the presence of idiopathic pulmonary fibrosis at death. Respir Med. 2012;106:588-93.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic pulmonary fibrosis: A study of 46 patients from Western India: Clinical presentations and survival. Turk Thorac J. 2015;16:114-20.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431-40.
- [CrossRef] [PubMed] [Google Scholar]
- Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:538-42.
- [CrossRef] [PubMed] [Google Scholar]




