
Translate this page into:
Pediatric interstitial lung disease
*Corresponding author: Andrew Bush, Department of Paediatric Respiratory Medicine, Imperial College and Royal Brompton Hospital, London, Middx, United Kingdom. a.bush@rbht.nhs.uk
-
Received: ,
Accepted: ,
How to cite this article: Bush A, Gilbert C, Gregory J, Nicholson AG, Semple T, Zampoli M, et al. Pediatric interstitial lung disease. J Pan Afr Thorac Soc 2021;2(1):18-32.
Abstract
Interstitial lung disease in children (chILD) is rare and encompasses more than 200 entities, with new especially genetic causes being discovered. Several classifications have been proposed, and there is considerable overlap with entities which present in adult life. Presentation may be shortly after birth with acute respiratory distress and in infancy and childhood either with a primary respiratory presentation or with systemic symptoms such as poor feeding and failure to thrive. Newborn acute presentations are usually due either to a mutation in one of the surfactant protein (Sp) genes or the alveolar capillary dysplasia (ACD)-congenital alveolar dysplasia (CAD) spectrum. The latter usually progress rapidly to extracorporeal membrane oxygenation, and early lung biopsy is advisable to prevent prolonged futile treatment being offered. Outside the newborn period, a staged protocol for investigation is proposed. This starts with a computed tomography scan, which confirms or otherwise the presence of chILD, and occasionally can lead to a specific diagnosis. In particular in settings where there is a high burden of infection, infective mimics of chILD need to be excluded. The next investigations aim to try to move from pattern recognition to specific diagnoses, both genetic and environmental. The speed of progression to lung biopsy will depend on the clinical state of the child, and the biopsy itself may suggest a hunt for a new underlying cause, such as immunodeficiency. Specific genetic causing chILD includes mutations in SpB and SpC and processing genes (thyroid transcription factor-1 [TTF-1] and adenosine triphosphate-binding cassette subfamily A) (the last three can present at any time in the life course); genes involved in Sp catabolism (granulocyte-macrophage colony factor receptor A and B genes), an increasing number implicated in the ACD-CAD spectrum, and other non-Sp related genes such as Filamin-A and integrin genes. Environmental causes are also important and vary across the world. Vaping has been implicated as causing a large number of chILDs, and a vaping history is essential in any young person with an unusual respiratory illness. Medications, both prescribed and over-the-counter such as oily laxatives, are also causes of chILD. There are important conditions of unknown cause presenting in early childhood. Neuroendocrine cell hyperplasia of infancy (NEHI) and pulmonary interstitial glycogenosis generally have a good prognosis, and are probably best considered as part of a spectrum of pulmonary dysmaturity syndromes, in some of which underlying gene mutations have been detected, for example, TTF-1 for NEHI. Pulmonary alveolar proteinosis is an example of an umbrella description, which may present at any age, and has a number of underlying causes with different specific treatments, underscoring the need to move from pattern recognition to specific diagnoses. chILDs have important implications for adult physicians; there may be late as yet poorly described sequelae of the disease or its treatment in adult life; there may be genetic implications for the wider family; and there may be late chILD relapses. Smooth transition to adult services is essential for all chILD survivors, with pediatric and adult chest physicians working closely together.
Keywords
Alveolar capillary dysplasia
Congenital alveolar dysplasia
Hypersensitivity pneumonitis
Lipoid pneumonia
Neuroendocrine cell hyperplasia of infancy
Pulmonary alveolar proteinosis
Pulmonary interstitial glycogenosis
Surfactant protein gene

INTRODUCTION
Children’s interstitial lung disease (chILD) is an umbrella term comprising a group of more than 200 conditions, which is growing in number every day. There are numerous genetic, environmental, systemic inflammatory, and other causes. They are very rare (estimated at around one in 300,000 children),[1,2] and they differ in important ways from adult interstitial lung diseases. Notably, chILD has to be considered in the context of the developing respiratory tract, and alveolar growth disorders and pulmonary hypoplasia are part of many chILD disorders,[3] whereas adult ILD is encountered in the context of the fully developed respiratory tract. There are also very important global differences in the nature of chILD, something which is of international importance in the era of globalization. Furthermore, treatment of chILD, most notably systemic corticosteroids, may also affect alveolar growth and development in ways that are irrelevant to adult disease,[4] but highly relevant to adult follow-up of these patients.
The rarity of chILD (about two orders of magnitude less common than adult disease, with much greater diversity of pediatric diagnoses) means that no single center will see enough cases to develop expertise in all causes; typically, even a specialist center will see no more than five new cases/year.[5] This means that collaboration is essential, both in diagnostic pathways and treatment algorithms. There are several international collaborations across the world, and there is no substitute for multicenter, multidisciplinary team review of data, especially imaging, histopathology, and genetics.[6,7]
Numerous different chILD classifications have been proposed,[8-10] which have moved the field forward; it is arguable that these have now become unwieldy as more and more entities have been delineated. Conventional classifications divide chILD by age of onset: 0–2 and over 2 years of age. However, there are many entities common to both age brackets, and perhaps, it is more logical (and clinically useful) to consider conditions presenting acutely at birth, and those with later, often more indolent, presentations (respiratory and non-respiratory). Of course even this classification is imperfect; there are conditions (e.g., surfactant protein [Sp] C and adenosine triphosphate [ATP]-binding cassette subfamily A member 3 [ABCA3] mutations) which can present at any age. Furthermore, causes of chILD will differ across the world, particularly environmental causes. It is essential, particularly in a lowand middle-income country (LMIC) setting, to determine locally important causes of chILD, a topic to which we return later in this article.
Finally, we need to consider our targets in understanding chILD. Cystic fibrosis (CF) is perhaps the best paradigm for moving from reactive, non-specific treatments such as antibiotics for infection and prednisolone for inflammation, to direct, proactive targeting of the basic defect. The key in CF was identifying the gene and then the subclasses of genetic abnormalities (I–VII).[11] The result was identification of specific molecules targeted at the disparate molecular defects, such as Ivacaftor[12] and Kaftrio[13,14] which have resulted in huge clinical benefits. There are increasing numbers of specific therapies for chILD, meaning that very specific diagnoses should be our target. These are not only genetic but also environmental where elimination of a specific insult, be it toxic or allergic, may be curative.
PRESENTATION OF CHILD
The most dramatic presentation is in the newborn period, with acute and unexpected severe respiratory distress in a term baby. Later, presentations are usually subacute or chronic, but there are exceptions, such as acute hypersensitivity pneumonitis (HSP). Presentation later in life is with either or both of respiratory (tachypnea, increase in work of breathing, and going blue) and non-respiratory (poor feeding and failure to thrive) manifestations. Each group will be considered in turn. Finally, there will be detailed consideration of selected individual conditions.
Newborn acute presentation of chILD
Although chILD can of course be seen in children born prematurely, it is rare compared to other causes of respiratory compromise, especially lung disease of prematurity.[15] Typically, the baby is born at term, needs immediate ventilatory support for no obvious reason, and is transferred to neonatal intensive care. The lungs are stiff, and ventilatory requirements are high. The chest radiograph (CXR) shows diffuse ground glass shadowing, as does the computed tomography (CT) scan (if performed). The course is either chronic and stable or rapidly deteriorating, with the baby either remaining on high pressure, high fractional inspired oxygen (FiO2)-positive pressure ventilation (IPPV), or deteriorating and being placed on extracorporeal membrane oxygenation (ECMO). One of the Sp gene mutations is likely the underlying cause of those running a chronic stable course, and the alveolar capillary dysplasia (ACD)-congenital alveolar dysplasia (CAD) spectrum causative of the rapidly deteriorating phenotype progressing to ECMO. In these desperately tragic circumstances, ideally, a rapid genetic diagnosis should be sought. This may not be possible because (a) genetic testing is not available, or results will be long delayed (e.g., ACD) wherein many mutations are in the promoter regions of the FOXF1 gene, not in an exon);[16] (b) rapid genetic testing is uninformative (some cases of biopsy proven Sp-B disease). In such cases, the question of the risks and benefits of lung biopsy arises. Our own view is that if a baby with an undiagnosed respiratory condition requires ECMO, a lung biopsy should be performed. This would be expected to be diagnostic of the ACD-CAD spectrum and allow withdrawal of what would be futile treatment in the case of ACD. There are concerns about the risks of bleeding during biopsy on an anticoagulated child, but in fact there is wide experience of safely performing biopsy both on ECMO[17,18] and also on cardiopulmonary bypass.[19-21] The question of when to biopsy a child who is stable but on high pressure, high FiO2 IPPV is more difficult, but if genetic studies are negative and the child is not improving, we would advocate lung biopsy to assess the likelihood of a reversible condition. Some of these conditions are discussed in more detail below and in a recent review.
chILD presenting outside the newborn period
The presenting symptoms (above) are non-specific, as are the physical signs. There may be digital clubbing, cyanosis, recession, crackles, and/or wheeze. The CXR is almost inevitably non-specific or may even appear normal, so detailed further investigation is essential. The diagnostic pathway may vary depending on the resources available. If chILD is suspected, the usual next step is a CT scan, to confirm or refute this possibility; however, circumstances may preclude moving a sick, ventilated child to the scanner, and it may be better under some circumstances, especially in LMIC, to proceed to invasive testing without a CT. If the HRCT suggests that the diagnosis is not a diffuse lung disease, then this is investigated on standard lines. Otherwise, a scheme of investigation, updated from the chILD-EU proposals,[22] is given in [Figure 1]. The key target is to move from pattern recognition (e.g., neuroendocrine cell hyperplasia of infancy [NEHI] and desquamative interstitial pneumonia [DIP]) to specific endotypes and even subendotypes (above). The urgency of investigations, and specifically that of invasive tests such as lung biopsy, is driven by the clinical situation. In fact, the infant stable on low flow oxygen, with no worrying red flags,[23] may appropriately never in fact undergo lung biopsy.
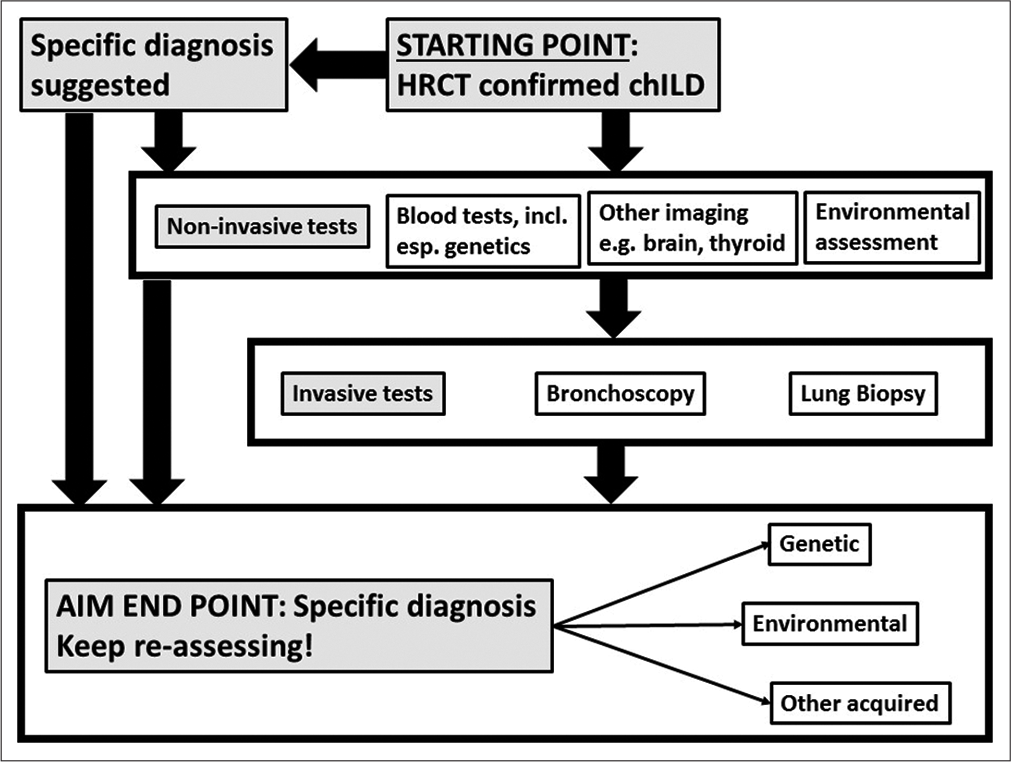
- Protocol for the investigation of suspected pediatric interstitial lung disease. There will to be regional differences in how it is applied in practice.
CT suggestive of a specific diagnosis examples [Figure 2] would include the typical right middle lobe and lingula ground-glass shadowing of NEHI; a mixture of cysts and nodules with upper lobe predominance pointing to Langerhans cell histiocytosis; soft centrilobular nodules suggestive of HSP; and the typical crazy paving appearance of pulmonary alveolar proteinosis (PAP). Lung biopsy would not be needed to confirm these diagnoses, but non-invasive testing, for example, for avian precipitins in the case of HSP and delineation of the specific causes of PAP would need to be undertaken (below).
CT confirms chILD, but no specific diagnosis possible a detailed history of any systemic features of the disease and potential environmental exposures is essential at this stage, if not already performed. Environmental exposures include potential drivers of HSP, medication history (both physician prescribed and over-the-counter), and especially vaping, which has been implicated in a wide range of acute, subacute, and chronic chILDs.[24-26] Blood tests especially focusing on relevant gene mutations and where appropriate, rheumatological investigations are performed. Consideration should be given as to other imaging, including echocardiography to exclude cardiac mimics of chILD and assess whether pulmonary hypertension is present. If a filamin A (FNLA) mutation is suspected, a brain magnetic resonance imaging (MRI) showing periventricular nodular hyperplasia will confirm the diagnosis. Brain-lung-thyroid syndrome due to a thyroid transcription factor-1 (TTF-1/NKX2.1) mutation could be suspected at this stage by finding abnormalities on thyroid function tests with abnormal movements (chorea).
Could this be a diffuse lung disease mimicking chILD? Infective mimics of chILD are an important diagnostic consideration. These may occur in the setting of a child with a known congenital or acquired immunodeficiency, including iatrogenic, or a child with a normal immune system but in a high infection setting. Examples would include opportunistic infection after chemotherapy for malignancy or organ transplant, and human immunodeficiency (HIV)-related infections. Miliary tuberculosis may mimic chILD in an otherwise normal child. In these contexts, bronchoalveolar lavage (BAL) (either bronchoscopic or blind in a ventilated child) may be diagnostic but, if it is not, rather than repeated lavages, progression to a lung biopsy is probably the wisest next step. In a case series from South Africa, in a population with a high prevalence of tuberculosis and HIV, high diagnostic yield was reported on biopsy, including new diagnoses of treatable infections, especially tuberculosis.[27] This series also underscores the folly of blind treatment of diffuse lung disease with high-dose corticosteroids in high infection burden settings, and the need to pursue a diagnosis aggressively. Furthermore, even in high burden settings, non-infective chILD may be diagnosed.
Are invasive tests indicated? The decision whether to proceed to further testing must be made on an individual basis. If the child is stable, it is reasonable to wait for the results of non-invasive tests before reaching a decision about invasive testing. Indeed, if the child is well and stable, and clinically a period of observation without giving potentially toxic treatment such as pulsed methylprednisolone is indicated, it may be appropriate not to do any further testing. If a diagnosis needs to be pursued, unless it is thought that a bronchoscopy and BAL will be diagnostic without a lung biopsy being performed (and this is usually only for infective mimics of chILD), it is best to proceed to a combined lung biopsy and bronchoscopy and BAL under the same anesthetic. Pulmonary hemorrhage can certainly be diagnosed on BAL, but many would advocate for a lung biopsy to look for an underlying pulmonary vasculitis in this context. Although transbronchial biopsy is very useful in the context of lung transplantation, it is not without risk, and the tissue samples are usually too small to be diagnostic in chILD. The exception may be very specific entities such as pulmonary alveolar microlithiasis.[28] The biopsy should be planned, with close collaboration between the physician requesting the biopsy, the pathologist and the surgeon, so the opportunity is not wasted.[22] Biopsy sites may be targeted, dependent on the imaging. Samples should be collected and ideally sent fresh to the laboratory, with separate samples sent to microbiology if appropriate. Once there, samples can be taken for potential electron microscopy and then fixed, ideally through gentle inflation of the parenchyma with formalin elec. Once the biopsy has been reported, a detailed reevaluation of the need for further tests is undertaken. For example, there may be findings suggestive of immunodeficiency [Table 1]. Pulmonary vasculitis and abnormalities in multiple lung compartments (airway, parenchyma, and vascular) suggest a multisystem disease, and a rheumatological consultation and further serological testing [Table 2] may be appropriate.
The hoped for end-point: Finding a specific genetic or environmental cause of chILD, as more specific treatments become available, a precise diagnosis where possible becomes ever more important. In the USA chILD Registry in 2019, there were 415 cases enrolled of whom 180 (43.5%) had undergone lung biopsy. Of these, 51 (28.3%, 12% of the total) also had informative genetics (Lisa Young, personal communication). It is likely that fewer biopsies will be being done in the future, as more gene mutations are implicated in chILD, and this will hopefully lead to novel, more specific therapies.
What are the options for non-specific treatment and what are the results? There are no published randomized controlled trials of any treatment for chILD. European treatment protocols have been published.[6] In summary, combinations of corticosteroids (either oral prednisolone or pulsed methylprednisolone, the latter can be combined with daily or alternate day prednisolone) and azithromycin and hydroxychloroquine, the latter two for their potential immunomodulatory and anti-fibrotic effects. In terms of prognosis,[29] response was better in those age ≥6 months and with oxygen saturations ≥94%.
For the group as a whole, response was seen within 3 months of starting treatment. In terms of duration of treatment, a 3-month initial trial would be reasonable. Thereafter, further treatment should depend on the response and be assessed on an individual basis.
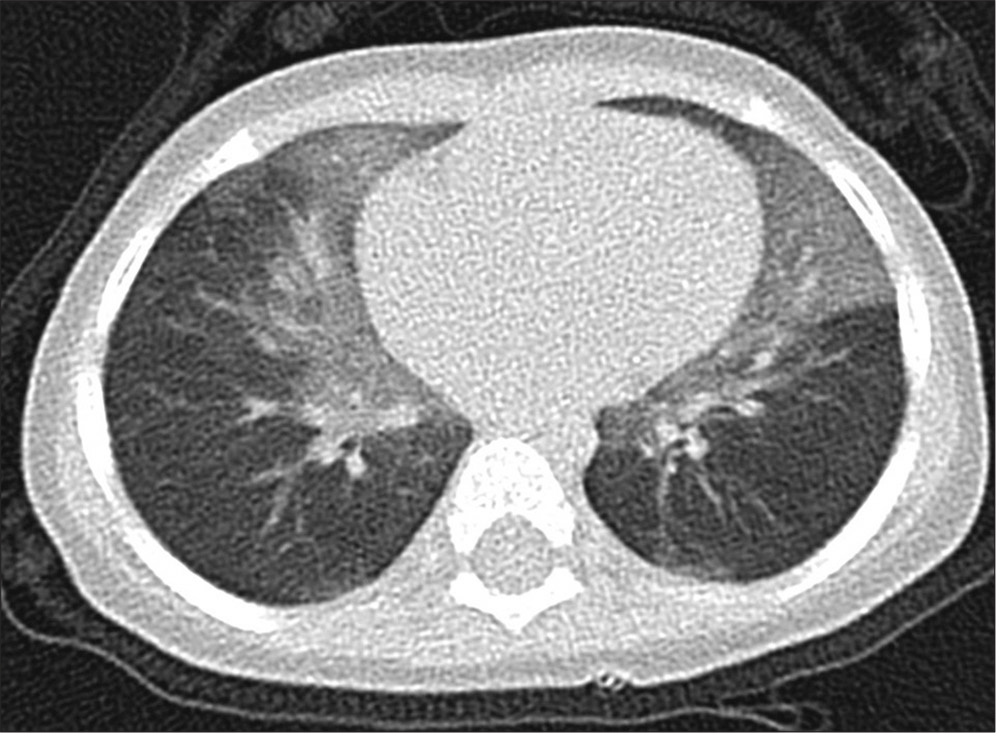
- A 12-month-old girl with right middle lobe and lingula lobe predominant ground-glass opacification, consistent with her clinical features of neuroendocrine cell hyperplasia of infancy.
| Biopsy diagnosis | Pathological features |
|---|---|
| Cellular bronchiolitis | Chronic peribronchial inflammation |
| Follicular bronchiolitis | Hyperplasia of the bronchial associated lymphoid tissue, or bronchiolar nodular lymphoid hyperplasia, leads to lymphoid follicles with germinal centers in the distal airway wall |
| Cellular non-specific interstitial pneumonia | Chronic inflammation of the interstitium with no evidence of fibrosis |
| Granulomatous lymphocytic interstitial lung disease | Granulomatous inflammation and proliferative lymphocytic abnormalities |
| LIP | Polyclonal infiltration of the interstitium and alveolar spaces with lymphocytes and plasma cells |
| Lymphoproliferative disease | Monoclonal lymphocytic infiltrates |
LIP: Lymphoid interstitial pneumonia
| Serology | Rheumatological differential diagnosis |
|---|---|
| Anti-nuclear antibody | Normal child Systemic lupus erythematosus Oligoarticular juvenile chronic arthritis Juvenile dermatomyositis Systemic sclerosis |
| Anti-double-stranded DNA | Mixed connective tissue disease Systemic lupus erythematosus |
| Anti-Sm | Systemic lupus erythematosus |
| Anti-Ro/La | Systemic lupus erythematosus Sjogren’s syndrome |
| Anti-ribonucleoprotein | Systemic lupus erythematosus Mixed connective tissue disease Systemic sclerosis |
| Anti-cardiolipin | Systemic lupus erythematosus Anti-phospholipid antibody syndrome |
| Anti-Jo-1 | Myositis |
| Anti-Scl-70 | Systemic sclerosis |
| Anti-centromere | Limited Systemic sclerosis |
| ANCA • PR3 • MPO |
Vasculitides GPA MPA |
| Rheumatoid factor | Rheumatoid factor positive juvenile chronic arthritis |
ANCA: Antineutrophil cytoplasmic antibody, GPA: Granulomatosis with polyangiitis, MPA: Microscopic polyangiitis, MPO: Myeloperoxidase
SPECIFIC GENETIC CAUSES OF CHILD
Disorders of surfactant protein synthesis
Perhaps, the best known and studied are the disorders of Sp synthesis and catabolism. There are four Sps of which two (SpA and SpD), together with mannose-binding lectin, form the connecting family, part of the innate immune system.[30] SpB and SpC are surface active, and prevent alveolar collapse. Mutations in these genes cause interstitial lung disease. This tidy classification is not altogether valid, since ILD in adults is also caused by SpA mutations.[30] The SpB and SpC gene products undergo complex post-transcriptional and post-translational modification to form the mature proteins, and part of the process requires adenosine triphosphate-binding cassette 3 (ABCA3).[31] The expression of these three genes is controlled in part by thyroid TTF-1 (NKX2.1). Thus, there are four groups of genetic causes of disordered Sp synthesis, the features of which are summarized in [Table 3]. SpB mutations typically lead to severe neonatal onset chILD, leading to death or lung transplantation usually within a year of birth. Rare cases of partial deficiency with prolonged survival have been described.[32,33] SpC, TTF-1, and ABCA3 disease may present at any age. Babies with two severe ABCA3 mutations typically present at birth and are dead or transplanted by a year of age.[34] Those with one or two mild mutations may survive long term, with prolonged periods of stability. The disorders of Sp catabolism are summarized in [Table 4]. Physiologically, surfactant is mainly cleared by alveolar macrophages and requires the granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR). There is a small contribution of the mucociliary escalator. Mutations in the CSFR alpha (CSF2RA) and beta (CSF2RB) chains are the most common disorders of Sp clearance; the others listed in [Table 4] are very rare indeed.[35] Typically but not inevitably, CSFR mutations present later than outside the newborn period.[35] CSF2RA may be associated with Turner’s syndrome (46, X0), with a mutation in the single X chromosome.[36] Treatment options include whole lung lavage[37] and bone marrow or stem cell transplantation.
| Gene | SFPTB | SFPTC | ABCA3 | NKX2.1 |
|---|---|---|---|---|
| Chromosome | 2p11.2 | 8p21.3 | 16p13.3 | 14q13.3 |
| Protein | SpB | SpC | ABCA3 | TTF.1 |
| Phenotype | Severe neonatal RDS Occasional mild, partial forms |
Severe neonatal RDS Non-resolving RSV bronchiolitis chILD Adult IPF/UIP (NB different manifestations including age of onset in same family kindred) |
Neonatal RDS chILD |
Neonatal RDS, chILD Recurrent respiratory tract infection Hypothyroidism Neurological disease (chorea, ataxia) (Brain-lung-thyroid syndrome – not all present in individual) |
| Inheritance | Autosomal recessive >40 mutations, 121ins2 in two thirds. (30% no gene mutation found; diagnosis on immunostaining) |
Autosomal dominant >60 mutations, p.Ile173Thr in one-third. Sporadic; variable penetrance |
Autosomal recessive >200 mutations, Glu292Val 10%; large complex gene |
Autosomal dominant |
| Incidence | <1 in 1,000,000 | Unknown | 1 in 10,000–20,000 | Unknown |
| Course | Neonatal onset, rapidly progressive | Variable; neonatal onset cases may survive long term | 60% early death (especially if two severe mutations) Generally severe but can stabilize |
Variable |
| Treatment | Lung transplant | Supportive Consider steroids, hydroxychloroquine, azithromycin Lung transplant |
Supportive Consider steroids, hydroxychloroquine, azithromycin Lung transplant |
Supportive Consider steroids, hydroxychloroquine, azithromycin Lung transplant |
chILD: Childrens’ interstitial lung disease, RDS: Respiratory distress, RSV: Respiratory syncytial virus, Sp: Surfactant protein
| Gene | CSF2RA | CSF2RB | GATA2 | OAS1 | SLC7A7 | MARS |
|---|---|---|---|---|---|---|
| Chromosome | Xp22.33 and Yp11.2 | 22q2.1 | 3q21.3 | 12q24.13 | 14q1.2 | 12q13.3 |
| Protein function | GMCSF alpha receptor subunit | GMCSF beta receptor subunit | Transcription factor | Oligoadenylate synthesis | Amino acid transporter (Lysinuric protein intolerance) | Methionyl-tRNA synthetase |
| Inheritance | X-linked recessive | Autosomal recessive | Sporadic Autosomal dominant |
Sporadic Autosomal dominant |
Recessive | Recessive |
| Age of onset | Infancy to adult | Infancy to adult | Childhood | Early infancy | Infancy | Infancy to adult |
| Extrapulmonary involvement | Only if part of a large deletion | No | Leukocytopenias Malignancy Infection |
Hypogammaglobulinemia Recurrent infection |
Gastrointestinal Renal |
Hepatic Anemia Hypothyroid |
| Treatment | Whole lung lavage Stem cell transplant |
Whole lung lavage Stem cell transplant |
? | Consider stem cell transplant | ? Whole lung lavage useless |
? |
The alveolar capillary dysplasia-congenital alveolar dysplasia syndrome
This group of diseases has been reviewed in detail recently,[38] they comprise acinar dysplasia, CAD, pulmonary hypoplasia, and ACD. They all present at or soon after birth, and are generally rapidly fatal, although mild forms with more prolonged survival have been described.[39-42] [Table 5] summarizes these conditions, including relating them to phases of arrested lung development.[43] There is most recent interest in ACD. Historically, it was thought that there was coexistent misalignment of pulmonary veins but these are in fact normally positioned expanded bronchial veins, draining the parenchyma because the pulmonary veins are occluded[44,45] [Figure 3]. FOXF1 drives angiogenesis and endothelial cell proliferation by multiple different pathways. This is important in trying to determine treatments; more than 70 FOXF1 variants and 60 copy number variant (CNV) deletions involving the gene and/or its upstream regulatory region have been reported,[16] and the precise signaling pathways are unknown for many. Signal transducer and activator of transcription 3 (STAT3) levels and the STAT3 signaling pathway are reduced in at least some murine and human ACD lungs. In a novel study, a Foxf1WT/S52F mouse was generated using CRISPR technology (the homozygous deletion is embryo lethal).[46] Intravenous administration of STAT3 containing nanoparticles corrected the murine disease phenotype. The authors caution that this approach may not be applicable in all mutations. It may be that knowledge of ACD subendotypes will be needed in the future, similar to CF where knowledge of such subendotypes has been needed to target modulator treatment appropriately.
| Gene | TBX4 | FGF10 | FGFR2 | FOXF1 |
|---|---|---|---|---|
| Location | 17q31.1-2 | 5p.12 | 0q26.13 | 16q24.1 |
| Function | Transcription factor | Encodes a fibroblast growth factor protein | Receptor protein | Unknown |
| Inheritance | Autosomal recessive | Autosomal recessive | Autosomal recessive | Autosomal dominant |
| Disease phenotype | Acinar dysplasia Congenital alveolar dysplasia Childhood pulmonary hypertension |
Acinar dysplasia Congenital alveolar dysplasia | Acinar dysplasia | Alveolar capillary dysplasia with misaligned pulmonary veins |
| Putative phase of lung development | Acinar dysplasia: Pseudoglandular-canalicular Congenital alveolar dysplasia: Canalicular-saccular |
Acinar dysplasia: Pseudoglandular-canalicular Congenital alveolar dysplasia: Canalicular-saccular |
Acinar dysplasia: Pseudoglandular-canalicular | Canalicular-saccular-alveolar |
| Extrapulmonary manifestations | Skeletal | Skeletal Lacrimal and salivary glands |
Craniofacial syndromes | Cardiovascular Genitourinary Gastrointestinal (especially if part of large deletion) |
ACD: Alveolar capillary dysplasia, CAD: Congenital alveolar dysplasia
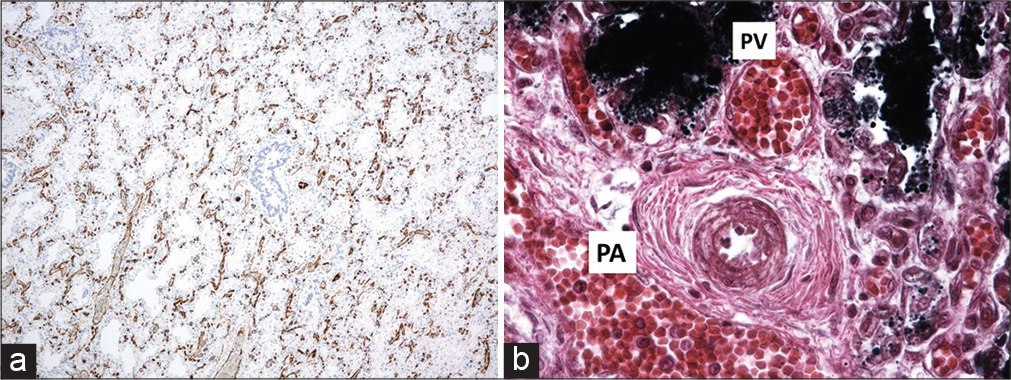
- Alveolar capillary dysplasia. (a) CD31 stain showing thickening of the interstitium and very little alignment between the very primitive alveolar spaces and the vasculature, (b) The pulmonary artery and vein is apposed in the bronchovascular bundle “Misaligned pulmonary veins.” Note the severe pulmonary hypertensive changes, with intimal proliferation and greatly increased medial muscle thickness.
Other genetic causes of chILD
This is an ever-expanding expanding list. Genetic causes of immunodeficiency and pulmonary vascular disease should also be considered, as more disease-specific treatments become available. Some recently genetic causes of chILD are summarized in [Table 6].
| Gene | FNLA | ITGA3 | COPA | TMEM173 | LRBA |
|---|---|---|---|---|---|
| Chromosome | Xq.28 | 17q21 | 1q23.2 | 5q31.2 | 4q31.3 |
| Protein function | Actin binding protein | Integrin; signaling cytoskeleton to extracellular matrix | Retrograde transport, Golgi to endoplasmic reticulum | STING Endoplasmic reticulum membrane signaling |
B-cell development and activation |
| Inheritance | X-linked dominant | Autosomal recessive | Sporadic Autosomal dominant |
Sporadic Autosomal dominant |
Autosomal Recessive |
| Age of onset | Neonatal to early infancy | Neonatal to early infancy | Infancy to adult | Infancy | Childhood |
| Pulmonary involvement | BPD-like phenotype Lung growth disorder Cystic disease |
RDS Growth disorder |
Alveolar hemorrhage chILD |
chILD | chILD LIP Bronchiectasis |
| Extrapulmonary involvement | CVS, CNS (periventricular heterotopia), skeletal | Renal (nephrotic syndrome) Skin, hair, nails (mild) |
Arthritis Renal Immune dysfunction |
Autoimmune vasculitis “SAVI” Skin |
CVID Auto-immunity Arthritis |
| Treatment | Lung transplant | None | Long term immune suppression Consider ruxolitinib |
Janus-associated tyrosine kinase inhibitors: Tofacitinib and ruxolitinib | Abatacept |
FNLA mutations
Inheritance is usually X-linked dominant, and there are prominent extrapulmonary manifestations including neurological, cardiovascular, and skeletal disease. The finding of periventricular nodular heterotopia on MRI is diagnostic of the condition. Pulmonary manifestations are largely of an alveolar growth disorder, with atelectasis combined with cysts and hyperinflation.[47-49] This mutation should be suspected if there is what appears to be atypical multifocal congenital lobar emphysema. There may also be tracheobronchomalacia related to reduce bronchial cartilage and pulmonary hypertension.
Integrin-α3 mutations
Integrin α3 is a transmembrane receptor mediating signaling between cells and their microenvironment, which forms heterodimers with integrin β1. Inheritance is autosomal recessive, and presentation is with early onset nephrotic syndrome, which is relatively mild, skin lesions, and a nail dystrophy mimicking epidermolysis bullosa, and chILD. The patients die of lung disease; CT scans show diffuse distortion of pulmonary architecture, coarsened interstitial changes, and septal thickening, and the lung histology is of irregular airspace expansion with preserved epithelium.[50] Diagnosis can be made genetically, or by finding absence of integrin α3 staining on a skin biopsy. Gain of function mutations leading to abnormal glycosylation of integrin α3 has also been described and is suspected if there is pathological glycogen staining on biopsy in integrin containing areas such as the reticular basement membrane.[51]
Coatomer subunit alpha mutations
At first sight, this looks like a rheumatological disease, with high titer autoantibodies (anti-nuclear antibody, anti-neutrophil cytoplasm antibody, and rheumatoid factor) associated with chILD (including alveolar hemorrhage) and inflammatory arthritis; there may also be renal disease. Presentation may be from infancy to young adult life.[52] Treatment is with long-term immunosuppression; the Janus activating kinase inhibitor ruxolitinib may also be effective.[53]
GATA-2 mutations
These mutations are mainly associated with malignancy and immune disease, but chILD including PAP has been described. GATA-2 associated PAP may be helped by whole lung lavage.[54-56]
TMEM173-STING (stimulator of interferon genes)
This is an early onset, multisystem disease, comprising any or all of rash, systemic inflammation, and peripheral vascular and tissue damage referred to as STING associated vasculopathy of infancy (SAVI). There are low titers of autoantibodies. The pulmonary phenotype is chILD with fibrosis, and hilar and paratracheal lymphadenopathy [Figure 4]. TMEM173 encodes STING; the disease is an interferonopathy caused by gain of function mutations in TMEM173.[57,58]
- A 6-year-old girl with a progressive interstitial lung disease, consisting of ground-glass opacification and cyst formation, subsequently found to have genetics consistent with stimulator of interferon genes.
LRB4 (lipopolysaccharide-responsive and beige-like anchor protein) mutations
The exact physiological role of LRB4 role is unknown, but it modulates cytotoxic T-lymphocyte-associated protein 4, also known as cluster of differentiation 152, which is an inhibitor expressed on activated T-cells and Tregs.[59] Common features include autoimmunity, lymphoproliferation, and immunodeficiency. Most patients are initially diagnosed as common variable immunodeficiency. Other manifestations include Type 1 diabetes, Burkitt cell lymphoma, and exocrine pancreatic insufficiency. The pulmonary manifestations are of chILD with dense lymphocytic infiltrates (including lymphocytic interstitial pneumonia). Importantly, beneficial effects have been reported with three drugs approved in other clinical contexts; abatacept (rheumatoid arthritis), belatacept (renal transplant rejection), and ipilimumab (immunotherapy of melanoma), with dramatic improvements in lung function and HRCT scan appearances.
SPECIFIC ENVIRONMENTAL CAUSES OF CHILD
chILD due to environmental toxins
The importance of the environment was underscored by the outbreak of a new chILD from Korea. There were 138 cases between 2006 and 2011, with increasing annual incidence.[60] The disease was seen in both children and adults, chILD presentation was at a mean age of 30.4 months, with rapidly progressive cough and dyspnea. There was no response to treatment. Imaging was non-specific, showing diffuse or centrilobular ground-glass opacities, patchy consolidation; air leaks were common. Lung biopsies showed bronchiolar injury and a bronchiolocentric acute lung injury pattern. The distal airway lesion varied from mononuclear cell infiltration to necrotizing and obliterative bronchiolitis. There was predominantly centrilobular diffuse alveolar damage. Accumulation of foamy histiocytes in peribronchial alveolar spaces was another feature. There were also features of an organizing pneumonia. Mortality was high (80/158, 58%). The cause was a humidifier disinfectant and, when this was withdrawn, no further cases occurred. It is difficult to believe that there will not be further outbreaks of environmental toxin-related lung disease in the future.
HSP
In a high-income setting, classical triggers are moldy hay (Micropolysporium faeni) and pet birds, the latter being most important in children. Other sources of bird allergens include feather bedding, and birds nesting in unused chimneys. Presentation in children is usually subacute or chronic, with progressive cough and breathlessness. The CT typically shows soft centrilobular nodules, and the diagnosis can be confirmed by determining serum IgG precipitins to the antigen.[61-63] Treatment is removal of the source of antigen and systemic corticosteroids. If an antigen cannot be identified, the rare possibility of telomere gene mutations should be considered.[64,65]
chILD differences across the world
It is very clear that we still have a lot to learn about the different environmental causes of chILD, and these will be very different across the world. The most systematic investigation of global variation in patterns of ILD has come from a collaboration between India and the USA.[66] The group reviewed 1084 adults seen in 27 centers in 19 Indian cities. There was a Central Indian Registry and independent diagnostic reviews in India and the USA. All patients had connective tissue disease serology performed, as well as spirometry and HRCT. Astonishingly, 47.3% had HSP, of which 48.1% related to air coolers. Other diagnoses include connective tissue disease associated ILD in 13.9% and idiopathic pulmonary fibrosis in 13.7%. The lesson for African investigators is that there is a need to identify the diseases that constitute African chILD, and how it differs from chILD in high-income settings, given the very different environments. One recent example was the apparent outbreak of chILD in the children of Zimbabwean immigrants to South Africa, which was due to lipoid pneumonia related to forcible administration of oily laxatives[67] [Figure 5].
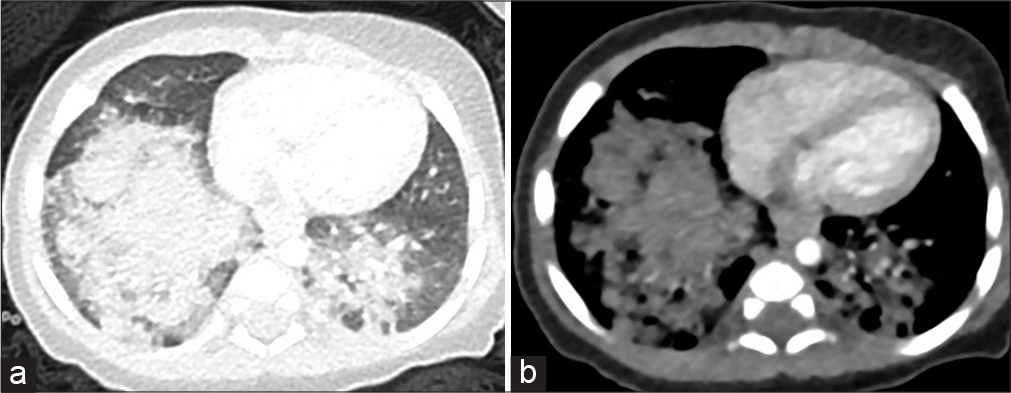
- A 2-month-old Zimbabwean boy with dense consolidation in the lower lobes and areas of low attenuation (best demonstrated on the soft-tissue window image) in keeping with exogenous lipoid pneumonia from oral administration of oils as prophylaxis against constipation. There is an important association with atypical infective organisms, particularly non-tuberculous mycobacteria, (a) Lung window, (b) Bone window.
The tobacco industry (yet again!)
Adult physicians have long recognized smoking associated ILD, but passive smoke exposure may also affect children. Respiratory bronchiolitis-associated interstitial lung disease is characterized by distal airway inflammation and interstitial lung disease. Symptoms are non-specific. Lung biopsy demonstrates clusters of pigmented macrophages.[68] Adult-type pulmonary Langerhans cell histiocytosis has been described in teenagers, although the relative contributions of active and passive smoke exposure may be difficult to determine. A much more potent cause of ILD in teenagers is vaping. Numerous different ILDs have been described, including acute eosinophilic pneumonia, lipoid pneumonia, alveolar hemorrhage, and organizing pneumonia.[69-72] A really important practice point is that exposure to vapes, or any other inhalational agent such as aerosols or butane,[73] should be sought in every child with any atypical lung disease.
Iatrogenic causes of chILD
Numerous medications have pulmonary toxicity, and a detailed medication history is mandatory. It is also important to consider whether non-prescription medications may be important, such as oily laxatives (above). The possibility of other, non-prescription remedies, for example, bush tea, as a cause of chILD should always be considered.
EXAMPLES OF OTHER CHILDS OF UNKNOWN CAUSE
NEHI
NEHI is another umbrella term, and the diagnosis should provoke the question “what sort of NEHI does this patient have?” Presentation is typically within a few weeks of birth, with tachypnea and increased work of breathing. Crackles may be audible, and the baby is usually oxygen dependent.[74] HRCT shows a very typical appearance, with ground-glass shadowing in the lingual and right middle lobes, and also in the perihilar regions[75] [Figure 2]. A recent study compared patients with “usual persistent tachypnea of infancy (PTI)” (typical NEHI, although the clinical term PTI is to be preferred to a pathological description when pathology is not available), aberrant PTI (atypical CT or clinical features), and controls.[23] There was overlap in NEHI cell numbers as identified on lung biopsy by bombesin staining between all three groups, and neither this nor any other paper on the subject has defined a robust normal range which can be used to diagnose NEHI. The authors recommended that lung biopsy only be performed if there are red flags, defined as onset age <4 weeks, unusual HRCT appearances, failure to thrive despite oxygen therapy, multisystem disease, and relentless deterioration. If a lung biopsy is performed, the histology is essentially normal, but staining for the neuropeptide bombesin shows increased numbers of positive cells and neuroepithelial bodies in the airways[76] [Figure 6]. These cells are expressed during normal lung development and are also found in other chILDs; they are not specific to NEHI.[77] There is no treatment, and the prognosis is good. In most cases, no underlying cause is found. However, a recent study measuring BAL proteins demonstrated that there were at least two endotypes of NEHI, clinically identical, but markedly different in protein expression.[78] Furthermore, a kindred with familial NEHI due to an underlying TTF-1/NKX2.1 mutation has been described.[79] Mutations in Forkhead box P1 (FOXP1) have been described in association with NEHI.[80] Undoubtedly, further genetic causes of NEHI will be described in the future.
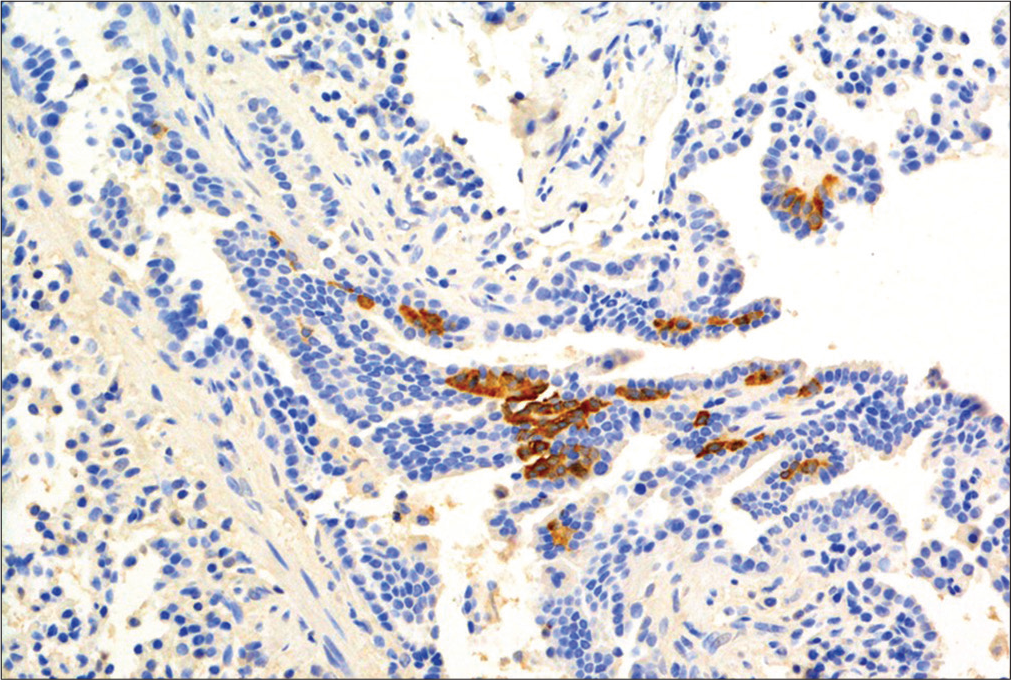
- Histology of neuroendocrine cell hyperplasia of infancy. There is obvious positive staining for bombesin-positive cells in the airway.
Pulmonary interstitial glycogenosis (PIG)
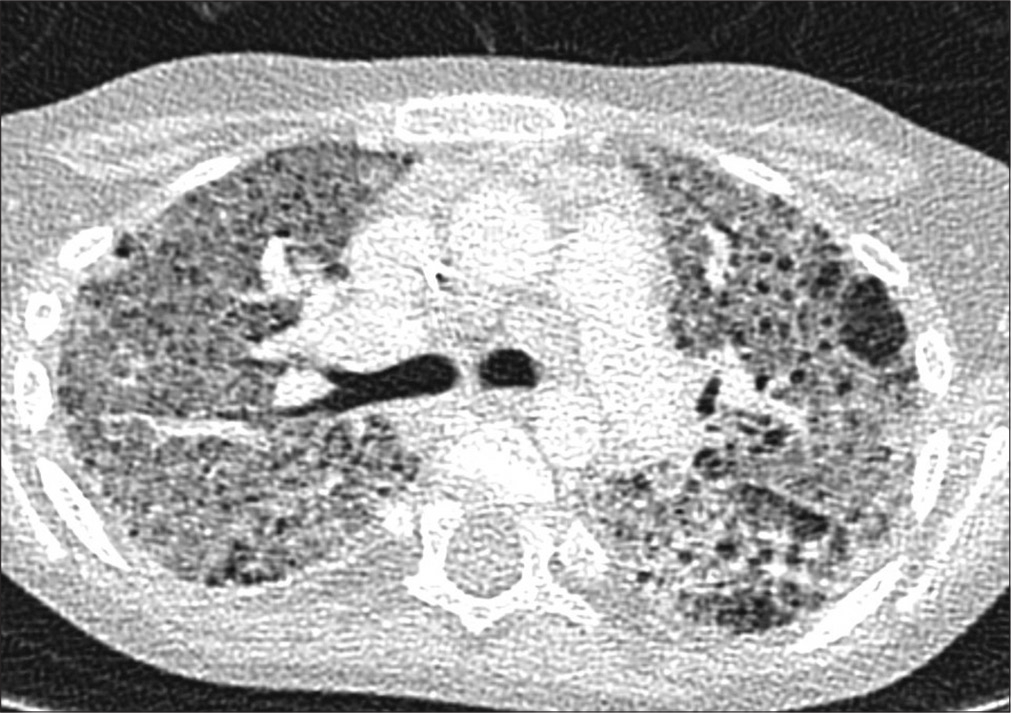
PIG typically presents non-specifically, with early onset respiratory distress. The CT scan is non-diagnostic, demonstrating that the lung architecture is distorted to a variable extent, with hyperinflated or hyperlucent regions with linear and ground-glass opacities. If a lung biopsy is performed, periodic acid–Schiff staining and electron microscopy confirm the presence of glycogen granules,[81] provided that the biopsy has been properly processed to prevent glycogen being leached out, underscoring the need for close consultation between surgeon and pathologist [Figure 7]. There are no established quantitative criteria for the pathological diagnosis of PIG. No other organ system is involved, and PIG is unrelated to any known glycogen storage disease. It is said that some cases are steroid responsive, the prognosis is good overall, but fatalities have been reported in preterm infants with PIG. As with NEHI cells, glycogen-positive cells are also found during normal fetal lung development. Furthermore, as with NEHI cells, PIG cells are also found in other contexts, for example, Hunter’s, Down’s, and Noonan’s syndrome. PIG cells are very commonly associated with alveolar growth disorders[8,82-84] and cardiac disease.[85,86] The developmental origin of PIG cells is still unclear, but they likely are of mesenchymal origin[87] and reflect a disorder of fibroblast differentiation.[87,88]
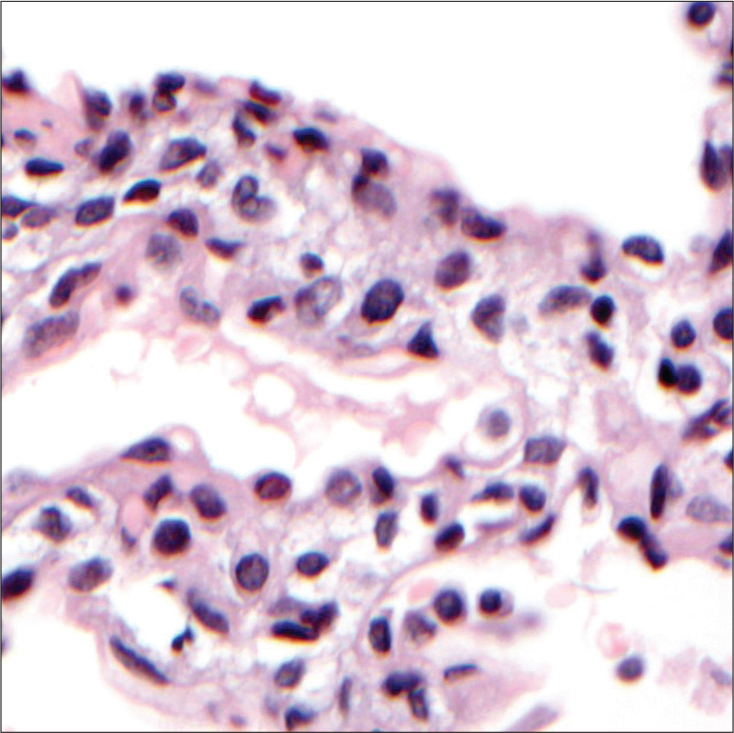
- Pulmonary interstitial glycogenosis. There is expansion of the interstitium, with vacuolated cells from which glycogen has been leached out during processing.
The spectrum of alveolar growth and dysmaturity syndromes
It is clear that NEHI and pig, and many of the alveolar growth disorders, are not discrete conditions but overlapping entities. We have proposed the umbrella term of “disorders of persistence of the fetal lung.”[89] These encompass any or all of persistence of the fetal airway (NEHI); persistence of the fetal interstitium (PIG); partial or complete alveolar growth failure; and abnormal growth of the lung vasculature. Within this spectrum, some specific diseases with known underlying genetics have been teased out, for example, acinar dysplasia (TBX4, FGF10, and FGFR2), CAD, (TBX4 and FGF10), alveolar-capillary dysplasia with misaligned pulmonary veins (FOXF1), and NEHI (TTF-1, FOXJ1, and above). Undoubtedly, more such endotypes and subendotypes will be discovered, but in the meantime, the approach that there are a spectrum of overlapping growth and developmental disorders which may affect multiple compartments and processes seems more useful that imagining that there are discrete, single compartment pathological entities.
PAP
It is usually easy to diagnose that the child has a condition fitting the umbrella term “PAP.” The HRCT shows background ground-glass shadowing with thickening of the secondary pulmonary lobules (“crazy paving”, [Figure 8]). Invasive testing is not necessary to confirm the diagnosis, but it is essential that the exact cause is determined [Table 7] because there are etiology-specific treatments. As can be seen, as with so many chILDs, the spectrum of etiologies is much wider than in the adult equivalent. Pediatric PAP has been recently reviewed.[90]
| Category | Underlying cause | Specific therapy? |
|---|---|---|
| Autoimmune | Ant-GMCSF autoantibodies | Whole lung lavage, inhaled or subcutaneous GMCSF |
| Disorders of surfactant protein metabolism | SpB, SpC, ABCA3, TTF-1 | Currently no specific treatments |
| Disorders of surfactant protein catabolism | GMCSFRA and GMCSFRB mutations | Stem cell transplantation |
| Other genetic | MARS, GATA-2, COPA, STING | COPA: Ruxolitinib? STING: Janus-associated tyrosine kinase inhibitors, tofacitinib and ruxolitinib |
| Metabolic | Lysinuric protein intolerance, Niemann-Pick disease | Avoid futile whole lung lavages |
| Immune deficiency | Many, including HIV | Treat underlying immune deficiency. Including bone marrow transplantation |
![A newborn with the typical “crazy-paving” appearance of pulmonary alveolar proteinosis (pulmonary alveolar proteinosis [PAP], diffuse ground-glass opacification with superadded interlobular septal thickening). The lack of pleural effusions shifts diagnostic suspicion away from the common mimics of PAP such as edema and lymphangiectasis/lymphangiomatosis. This child was later found to have a mutation in the gene coding an alpha subunit of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor (GM-CSFR2A).](/content/124/2021/2/1/img/JPATS-2-018-g008.png)
- A newborn with the typical “crazy-paving” appearance of pulmonary alveolar proteinosis (pulmonary alveolar proteinosis [PAP], diffuse ground-glass opacification with superadded interlobular septal thickening). The lack of pleural effusions shifts diagnostic suspicion away from the common mimics of PAP such as edema and lymphangiectasis/lymphangiomatosis. This child was later found to have a mutation in the gene coding an alpha subunit of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor (GM-CSFR2A).
chILD: LIFELONG IMPLICATIONS
There are very few longitudinal data about chILD and the long-term outlook, and there are many reasons why this needs to be rectified.[91]
The disease may recrudesce. Some, for example, SpC mutations, are known to run a relapsing and remitting course,[92] but the natural history of many chILDs is simply not understood.
The rarity of child conditions and their unknown natural history also severely limits the design of interventional trials of existing and novel therapies because of, for example, difficulties matching intervention patients with controls, differentiating which changes in the treatment arm are due to the intervention rather than the natural cause of the underlying disease, and determining the length of intervention required to detect a true effect.
There may be as yet poorly characterized sequelae of the disease. An example is the obstructive airway disease described in NEHI survivors in one small series.[93] Failure to appreciate the previous history may result in such patients being inappropriately treated with asthma medications. We have seen at least one long-term non-smoking adult who presented in infancy with a SpC mutation die in adult life of lung cancer, and there are other anecdotal cases of late malignancy in Sp mutations (Nogee L, personal communication).
Lung growth may be abnormal, both as a result of chILD associated pulmonary hypoplasia and also treatment, especially prednisolone. Failure to attain normal lung growth is an important risk factor for adult lung disease, including COPD.[94]
There may be genetic implications both for the patient and the wider family. As increasing numbers of specific genetic diagnoses are being made, what in childhood was thought to be idiopathic may in fact turn out to be a specific genetic endotype with new implications.
Finally, patient surveys have highlighted the extreme anxiety families go through when dealing with chILD, including related to not knowing the prognosis, the disruption of family life by acute exacerbations, and the need for oxygen and other equipment in the home.[95] The long-term scars left by these factors are not studied, but likely to be considerable. International chILD charitable and support organizations can help aid with social connectivity and emotional support. However, since these connections are online, low-income families who are more likely to experience digital divide, may be at a greater disadvantage to receiving this external support. Encouragement for families to reach out to chILD-related organizations is recommended.
One difficulty is ensuring a smooth transition to adult services, because such a transition is a rare event, and there are few if any child-specific transition pathways. There is also a major lack of registries that span developmental silos, and it is essential this is rectified.
SUMMARY AND CONCLUSIONS
We are entering a new age of chILD, in which specific genetic and environmental diagnoses, with specific treatments, are replacing the old approach of morphological descriptions. Pursuing diagnoses may be an ongoing process, and it is essential to keep revisiting diagnostic possibilities in a patient with an undiagnosed chILD, not least because of possible new treatments and implications for the wider family. Furthermore, chILD has an international dimension. The challenge to young investigators across the globe is to identify country-specific causes of chILD. Collaboration between groups is essential; clinicians, radiologists, pathologists, and geneticists need to assess cases, and the use of international consortia to broaden expertise will become ever more important. Finally, never forget the multiple different chILDs that can be caused by Vaping – if you see a respiratory case which is unusual, Cherchez la Vape!
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Idiopathic interstitial pneumonitis in children: A national survey in the United Kingdom and Ireland. Pediatr Pulmonol. 2002;34:23-9.
- [CrossRef] [PubMed] [Google Scholar]
- The incidence of interstitial lung disease 1995-2005: A Danish nationwide population-based study. BMC Pulm Med. 2008;8:24.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse lung disease in infancy and childhood: Expanding the chILD classification. Histopathology. 2013;63:743-55.
- [CrossRef] [PubMed] [Google Scholar]
- Triamcinolone-induced structural alterations in the development of the lung of the fetal rhesus macaque. Am J Obstet Gynecol. 1984;148:203-15.
- [CrossRef] [Google Scholar]
- Childhood interstitial lung disease: A systematic review. Pediatr Pulmonol. 2015;50:1383-92.
- [CrossRef] [PubMed] [Google Scholar]
- Research in progress: Put the orphanage out of business. Thorax. 2013;68:971-3.
- [CrossRef] [PubMed] [Google Scholar]
- Gilbert C, Schwerk N, ChILDEU Clinical Research Collaboration Management Committee, Members of the ChILDEU Clinical Research Collaboration Management Committee. The European research collaboration for children's interstitial lung disease (ChILDEU) ERS clinical research collaboration. Eur Respir J. 2018;52:1801855.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse lung disease in young children: Application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120-8.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse lung disease in biopsied children 2 to 18 years of age, Application of the chILD classification scheme. Ann Am Thorac Soc. 2015;12:1498-505.
- [CrossRef] [PubMed] [Google Scholar]
- Incidence and classification of pediatric diffuse parenchymal lung diseases in Germany. Orphanet J Rare Dis. 2009;4:26.
- [CrossRef] [PubMed] [Google Scholar]
- Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4:662-74.
- [CrossRef] [Google Scholar]
- A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-72.
- [CrossRef] [PubMed] [Google Scholar]
- VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379:1599-611.
- [CrossRef] [PubMed] [Google Scholar]
- Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809-19.
- [CrossRef] [PubMed] [Google Scholar]
- Respiratory distress in the newborn. Pediatr Rev. 2014;35:417-28.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical, histopathological, and molecular diagnostics in lethal lung developmental disorders. Am J Respir Crit Care Med. 2019;200:1093-101.
- [CrossRef] [PubMed] [Google Scholar]
- Open lung biopsy in pediatric patients on extracorporeal membrane oxygenation. J Pediatr Surg. 1996;31:1376-8.
- [CrossRef] [Google Scholar]
- Open lung biopsy in neonatal and paediatric patients referred for extracorporeal membrane oxygenation (ECMO) Thorax. 2004;59:328-33.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation. 1984;69:655-67.
- [CrossRef] [PubMed] [Google Scholar]
- Lung biopsy with frozen section as a diagnostic aid in patients with congenital heart defects. Am J Cardiol. 1981;47:77-84.
- [CrossRef] [Google Scholar]
- Lung biopsy in congenital heart disease: A morphometric approach to pulmonary vascular disease. Circulation. 1978;58:1107-22.
- [CrossRef] [PubMed] [Google Scholar]
- European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015;70:1078-84.
- [CrossRef] [PubMed] [Google Scholar]
- Persistent tachypnea of infancy, Usual and aberrant. Am J Respir Crit Care Med. 2016;193:438-47.
- [CrossRef] [PubMed] [Google Scholar]
- Life-threatening bronchiolitis related to electronic cigarette use in a Canadian youth. CMAJ. 2019;191:E1321-31.
- [CrossRef] [PubMed] [Google Scholar]
- An update on controversies in E-cigarettes. Paediatr Respir Rev. 2020;36:75-86.
- [CrossRef] [PubMed] [Google Scholar]
- Vaping and E-cigarette use, Mysterious lung manifestations and an epidemic. Curr Opin Immunol. 2020;66:143-50.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosing diffuse lung disease in children in a middle-income country: The role of open lung biopsy. Int J Tuberc Lung Dis. 2017;21:869-74.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary alveolar microlithiasis in childhood: Diagnosis by transbronchial biopsy. Pediatr Pulmonol. 1996;21:62-4.
- [CrossRef] [Google Scholar]
- One-year outcomes in a multicentre cohort study of incident rare diffuse parenchymal lung disease in children (ChILD) Thorax. 2019;75:172-5.
- [CrossRef] [PubMed] [Google Scholar]
- SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2015;192:1249-52.
- [CrossRef] [PubMed] [Google Scholar]
- ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296-303.
- [CrossRef] [PubMed] [Google Scholar]
- Delayed presentation and prolonged survival of a child with surfactant protein B deficiency. J Pediatr. 2017;190:268-70.e1.
- [CrossRef] [PubMed] [Google Scholar]
- Prolonged survival in hereditary surfactant protein B (SP-B) deficiency associated with a novel splicing mutation. Pediatr Res. 2000;48:275-82.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 2014;189:1538-43.
- [CrossRef] [PubMed] [Google Scholar]
- Whole-genome sequencing of a family with hereditary pulmonary alveolar proteinosis identifies a rare structural variant involving CSF2RA/CRLF2/IL3RA gene disruption. Sci Rep. 2017;7:43469.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med. 2008;205:2711-6.
- [CrossRef] [PubMed] [Google Scholar]
- Successful haematopoietic stem cell transplantation in a case of pulmonary alveolar proteinosis due to GM-CSF receptor deficiency. Thorax. 2018;73:590-2.
- [CrossRef] [PubMed] [Google Scholar]
- Interstitial lung disease in infancy. Early Hum Dev. 2020;150:105186.
- [CrossRef] [PubMed] [Google Scholar]
- Late presentation of alveolar capillary dysplasia in an infant. Pediatr Crit Care Med. 2006;7:177-9.
- [CrossRef] [PubMed] [Google Scholar]
- Prolonged survival in alveolar capillary dysplasia syndrome. Eur J Pediatr. 2004;163:181-2.
- [CrossRef] [PubMed] [Google Scholar]
- Nitric oxide therapy for an infant with PPH caused by misalignment of pulmonary veins with alveolar capillary dysplasia. J Pediatr Surg. 1997;32:99-100.
- [CrossRef] [Google Scholar]
- A late presenter and long-term survivor of alveolar capillary dysplasia with misalignment of the pulmonary veins. Eur J Pediatr. 2015;174:1123-6.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic disorders influencing lung formation and function at birth. Hum Mol Genet. 2004;3:R207-15.
- [CrossRef] [PubMed] [Google Scholar]
- Intrapulmonary vascular shunt pathways in alveolar capillary dysplasia with misalignment of pulmonary veins. Thorax. 2015;70:84-5.
- [CrossRef] [PubMed] [Google Scholar]
- Three-dimensional reconstruction identifies misaligned pulmonary veins as intrapulmonary shunt vessels in alveolar capillary dysplasia. J Pediatr. 2014;164:192-5.
- [CrossRef] [PubMed] [Google Scholar]
- Postnatal alveologenesis depends on FOXF1 signaling in c-KIT+ endothelial progenitor cells. Am J Respir Crit Care Med. 2019;200:1164-76.
- [CrossRef] [PubMed] [Google Scholar]
- Filamin A (FLNA) mutation-A newcomer to the childhood interstitial lung disease (ChILD) classification. Pediatr Pulmonol. 2017;52:1306-15.
- [CrossRef] [PubMed] [Google Scholar]
- A review of filamin A mutations and associated interstitial lung disease. Eur J Pediatr. 2019;178:121-9.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital emphysematous lung disease associated with a novel Filamin A mutation. Case report and literature review. BMC Pediatr. 2019;19:86.
- [CrossRef] [PubMed] [Google Scholar]
- Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012;366:1508-14.
- [CrossRef] [PubMed] [Google Scholar]
- Intronic ITGA3 mutation impacts splicing regulation and causes interstitial lung disease, nephrotic syndrome, and epidermolysis bullosa. Invest Dermatol. 2016;136:1056-9.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic causes and clinical management of pediatric interstitial lung diseases. Curr Opin Pulm Med. 2018;24:253-9.
- [CrossRef] [PubMed] [Google Scholar]
- Use of ruxoliitinib in COPA syndrome manifesting as life-threatening alveolar haemorrhage. Thorax. 2019;75:92-5.
- [CrossRef] [PubMed] [Google Scholar]
- Hematopoietic stem cell transplantation for pulmonary alveolar proteinosis associated with primary immunodeficiency disease. Int J Hematol. 2018;107:610-4.
- [CrossRef] [PubMed] [Google Scholar]
- GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med. 2015;15:87.
- [CrossRef] [PubMed] [Google Scholar]
- GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809-21.
- [CrossRef] [PubMed] [Google Scholar]
- Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. 2019;130:1669-82.
- [CrossRef] [PubMed] [Google Scholar]
- Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507-18.
- [CrossRef] [PubMed] [Google Scholar]
- Autoimmune disease, Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349:436-40.
- [CrossRef] [PubMed] [Google Scholar]
- Humidifier disinfectant-associated children's interstitial lung disease. Am J Respir Crit Care Med. 2014;189:48-56.
- [Google Scholar]
- Hypersensitivity pneumonitis in children. Curr Opin Pediatr. 2002;14:323-6.
- [CrossRef] [PubMed] [Google Scholar]
- Hypersensitivity pneumonitis in children: Clinical features, diagnosis, and treatment. Paediatr Drugs. 2005;7:235-44.
- [CrossRef] [PubMed] [Google Scholar]
- Bird fancier's lung: A state-of-the-art review. Clin Rev Allergy Immunol. 2012;43:69-83.
- [CrossRef] [PubMed] [Google Scholar]
- Rare protein-altering telomere-related gene variants in patients with chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2019;200:1154-63.
- [CrossRef] [PubMed] [Google Scholar]
- Telomeropathy in chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2019;200:1086-7.
- [CrossRef] [PubMed] [Google Scholar]
- Interstitial lung disease in India, Results of a prospective registry. Am J Respir Crit Care Med. 2017;195:801-13.
- [CrossRef] [PubMed] [Google Scholar]
- Exogenous lipoid pneumonia in children: A systematic review. Paediatr Respir Rev. 2020;33:45-51.
- [CrossRef] [PubMed] [Google Scholar]
- Respiratory Bronchiolitis-associated interstitial lung disease in childhood: New sequela of smoking. Pediatrics. 2015;136:e1026-9.
- [CrossRef] [PubMed] [Google Scholar]
- Life-threatening hypersensitivity pneumonitis secondary to E-cigarettes. Arch Dis Child. 2020;105:1114-6.
- [CrossRef] [PubMed] [Google Scholar]
- Acute eosinophilic pneumonia associated with non-cigarette smoking products: A systematic review. Adv Respir Med. 2020;88:142-6.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse alveolar haemorrhage secondary to E-cigarette vaping associated lung injury (EVALI) in a young European consumer. Eur Respir J. 2020;56:2000143.
- [CrossRef] [PubMed] [Google Scholar]
- Case report: Pulmonary interstitial fibrosis associated with inhalation exposure to aviation fuels. Int J Med Toxicol. 2001;4:20.
- [Google Scholar]
- Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol. 2005;40:157-65.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroendocrine cell hyperplasia of infancy (NEHI) Pediatr Radiol. 2006;36:1328.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest. 2011;139:1060-71.
- [CrossRef] [PubMed] [Google Scholar]
- Bombesin staining in neuroendocrine cell hyperplasia of infancy (NEHI) and other childhood interstitial lung diseases (chILD) Histopathology. 2015;67:501-8.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary aptamer signatures in children's interstitial and diffuse lung disease. Am J Respir Crit Care Med. 2019;200:1496-504.
- [CrossRef] [PubMed] [Google Scholar]
- Persistent lung disease in adults with NKX2.1 mutation and familial neuroendocrine cell hyperplasia of infancy. Ann Am Thorac Soc. 2016;13:1299-304.
- [CrossRef] [PubMed] [Google Scholar]
- FOXP1 haploinsufficiency: Phenotypes beyond behavior and intelectual disability? Am J Med Genet A. 2017;173:3172-81.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis: A new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med. 2002;165:1557-65.
- [CrossRef] [PubMed] [Google Scholar]
- Diffuse lung disease in infancy: A proposed classification applied to 259 diagnostic biopsies. Pediatr Dev Pathol. 2009;12:421-37.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: Radiographic and pathologic correlation. Pediatr Radiol. 2010;40:1562-5.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis associated with a spectrum of neonatal pulmonary disorders. Hum Pathol. 2017;68:154-65.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis: An unrecognized etiology of persistent pulmonary hypertension of the newborn in congenital heart disease. Pediatr Cardiol. 2013;34:1254-7.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis: A reversible underlying condition associated with D-transposition of the great arteries and severe persistent pulmonary hypertension. World J Pediatr Congenit Heart Surg. 2015;6:480-3.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary interstitial glycogenosis cells express mesenchymal stem cell markers. Eur Respir J. 2020;56:2000853.
- [CrossRef] [PubMed] [Google Scholar]
- Lipofibroblast phenotype in pulmonary interstitial glycogenosis. Am J Respir Crit Care Med. 2016;193:694-6.
- [CrossRef] [PubMed] [Google Scholar]
- Early onset children's interstitial lung diseases: Discrete entities or manifestations of pulmonary dysmaturity? Paediatr Respir Rev. 2018;30:65-71.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary alveolar proteinosis in children. Breathe (Sheff). 2020;16:200001.
- [CrossRef] [PubMed] [Google Scholar]
- Abandoning developmental silos: What can paediatricians and adult interstitial lung disease physicians learn from each other? Curr Opin Pulm Med. 2019;25:418-25.
- [CrossRef] [PubMed] [Google Scholar]
- Natural history of five children with surfactant Protein C mutations and interstitial lung disease. Pediatr Pulmonol. 2014;49:1097-105.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroendocrine cell hyperplasia of infancy: A prospective follow-up of nine children. Arch Dis Child. 2013;98:141-4.
- [CrossRef] [PubMed] [Google Scholar]
- Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med. 2015;373:111-22.
- [CrossRef] [PubMed] [Google Scholar]
- Childhood interstitial lung disease: Family experiences. Pediatr Pulmonol. 2015;50:1301-3.
- [CrossRef] [PubMed] [Google Scholar]




